
New World Screwworm: Fact Sheet & SHIC Article 6/10/2026 & USDA NWS Website
Register HERE for the August 4, 2026, New World Screwworm webinar (1 – 2:30 pm CT) & Find more information about webinar content here.
Project #: 24-076 | Principal Investigator: Igor Paploski | Institution: University of Minnesota | Posted: 7/17/2026 | Keywords: Swine, pathogens, biosecurity, PRRSV, PEDV, rendering, composting, dead-box, environment
This project evaluated if swine pathogens can be found around dead animal handling structures (dead boxes and composting bins) and if these areas may represent overlooked biosecurity risks on wean-to-market farms. We found that environmental contamination around dead boxes and composting areas was common, particularly on farms using rendering for mortality disposal. Farms utilizing rendering were more likely to have environmental samples test positive for PRRSV, PEDV, or PDCoV, and positive samples generally contained greater amounts of viral genetic material than those collected from composting sites. We also demonstrated that material present near dead animal handling areas can be mechanically moved by vehicles and personnel using a fluorescent marker as a proxy for contamination. Importantly, a simple intervention consisting of whitewash application around dead animal handling structures reduced the proportion of PRRSV-positive environmental samples from 13% to 3% within two days. Finally, our results suggest that mortality management practices and environmental conditions, such as recent rainfall or snowmelt, influence the amount of contamination found around these sites. Together, these findings highlight dead animal handling structures as an important biosecurity consideration and suggest that relatively simple management practices may help reduce environmental contamination and strengthen farm biocontainment and biosecurity.
Project #: 25-052 | Principal Investigator: Cesar A. Corzo | Institution: University of Minnesota | Posted: 6/23/2026 | Keywords: Monitoring, Swine Health, PRRS, PEDv, Senecavirus, Influenza, PRRSv 1H.18, Variants Under Monitoring
Objective 1: Monitor trends in pathogens incidence and prevalence – PRRSv, PEDv, PDCoV, Senecavirus viruses continued to be monitored, each maintaining historical patterns. However, the PEDv cumulative incidence trend was different in that a sharp increase was observed during the fall and into the winter. During 2025 we also explored and developed a method to estimate the breeding herd Influenza A virus (IAV) cumulative incidence. While we confirmed that it is possible to estimate the cumulative incidence based on VDL RT-PCR data, more work needs to be performed to avoid potential underestimation of this metric. Overall, the IAV cumulative incidence ranged between 1% and 5% over the past 10 years.
Objective 2: To conduct prospective monitoring of PRRSv sequence evolution and impact – During 2025 we continued to curate and expand our PRRSv ORF5 database. The representativeness of this database has enabled us to closely monitor throughout the year a newly emerged variant 1H.18. As this variant was monitored, a new system to monitor emerging variants was developed, Variants Under Monitoring (VUM) which is now shared with the public every month. The report is allowing producers to better understand the risk of every single variant that shows rapid dissemination characteristics. Since this report is based on the number of newly infected sites, the industry has an objective measure of variant fitness and thus risk. During 2025, we also assessed the possibility of sharing our weekly maps summarizing disease occurrence. While we believe this would be a great tool for the industry, we need to continue working on pathways to ensure producers and veterinarians feel comfortable with sharing such granular data.
Objective 3: To expand participation of producers to allow for all to be involved – During 2025 we made measurable progress toward broadening participation by successfully onboarding one additional production system, with two additional systems currently in the final stages of enrollment pending submission of required documentation. We have now accounted for 3.9M sows which approximately represents 65% of the U.S. breeding herd. With the goal of better understanding the health profile of the pigs entering the U.S. from Canada, some veterinary clinics and systems were approached to explore whether they would be interested in joining our voluntary project. While there was interest expressed, internal communications needed to occur before them making the decision to join. Fortunately, we did add one system that ships their weaned pig production into the Midwestern United States. On the other hand, we continue to work towards the characterization of each participating breeding herd from a filtration and mortality management perspective.
Project #: 24-022 | Principal Investigator: Wenjun Ma | Institution: University of Missouri | Posted: 6/19/2026 | Keywords: Swine, mammalian orthoreovirus, porcine adenovirus, prevalence, pathogenicity
Industry Summary: This project examined whether mammalian orthoreovirus (MRV) and porcine adenovirus (PAdV) are present in U.S. swine herds and whether these viruses can make pigs sick. These viruses have been reported in pigs in other countries, but little recent information is available for U.S. herds. Our goal was to give producers and veterinarians a clearer picture of how common these viruses may be and whether they pose a health risk. To answer these questions, we worked with veterinary diagnostic laboratories and collected samples from pigs in multiple states. We tested lung, intestinal, blood, and fecal samples for evidence of MRV and PAdV. We also developed a blood test for MRV antibodies to determine whether pigs had been exposed to this virus in the past. In addition, we carried out controlled pig studies to see whether MRV, PAdV, or both together could cause illness and spread to other pigs. The surveillance results showed very little direct evidence of active MRV infection in tissue samples: only three samples were positive for viral genetic material, and live virus could not be recovered from those samples. However, about 60% of blood samples were positive for MRV antibodies, suggesting that exposure to MRV is common in U.S. pigs even when active infection is not easily detected. For PAdV, no lung or intestinal tissue samples tested positive, but a small number of fecal samples were positive by PCR. Live virus could not be isolated from those samples either. Overall, these findings suggest that both viruses circulate in swine populations, with evidence pointing more toward intestinal shedding or prior exposure than widespread active disease detected in routine tissue submissions. In the pig studies, pigs infected with MRV, PAdV, or both developed mild clinical signs, including diarrhea and fever, and viral genetic material was found in intestinal tissues of infected pigs and contact pigs. Coinfection did not clearly worsen disease compared with infection by either virus alone based on clinical signs. For producers, these results suggest that MRV and PAdV can infect pigs and contribute to enteric illness, but they were not associated with severe or clearly distinct disease under the conditions tested in this study. These results are important to the swine industry because they show that MRV exposure is common and that both MRV and PAdV are capable of infecting pigs, especially in the intestinal tract. At the same time, the low detection rate of active virus in field samples is most likely due to sample limitations, including the small number of intestinal and fecal samples included in this study. Continued monitoring and surveillance is necessary to determine when these viruses matter most and whether they contribute to health problems when combined with other infections or management stressors. This information can help producers and veterinarians interpret diagnostic findings and make more informed herd health decisions.
Project #: 24-002 | Principal Investigator: Maria Sol Perez Aguirreburualde (P.I.) | Institution: University of Minnesota | Posted: 6/1/2026 | Keywords: Biosecurity, harvest plants, trailer contamination, PDCoV, PEDV, PRRSV, Senecavirus A
Industry Summary: Since 2018, African Swine Fever (ASF) has remained a persistent global threat, with continued spread and re-emergence across Asia, Europe, and the Americas. More recently, the introduction of Foot-and-Mouth Disease (FMD) in Europe, through two independent incursions, and the first-time detection of several FMD serotypes in new geographic regions have further demonstrated that transboundary animal diseases (TADs) are an evolving and intensifying concern. These events reinforce a core lesson first highlighted during the 2013 Porcine Epidemic Diarrhea Virus (PEDv) outbreak: the swine industry requires robust, real-time situational awareness to anticipate, prepare for, and respond to emerging threats.
Project #: 24-077 | Principal Investigator: Cesar A Corzo | Institution: University of Minnesota | Posted: 4/29/2026 | Keywords: Biosecurity, harvest plants, trailer contamination, PDCoV, PEDV, PRRSV, Senecavirus A
This project was conducted to better understand how important swine viruses spread at harvest facilities, with a particular focus on the unloading process. The study evaluated four major pathogens affecting the swine industry: Porcine Deltacoronavirus (PDCoV), Porcine Epidemic Diarrhea Virus (PEDV), Porcine Reproductive and Respiratory Syndrome Virus (PRRSV), and Senecavirus A (SVA). The main objectives were to assess whether contamination increases during unloading, and identify key factors such as season, cleaning practices, and driver behavior that influence contamination risk. To achieve these goals, sampling was conducted at a harvest facility every two weeks over a one-year period. During each visit, environmental samples were collected from three main locations: the dock, trailers upon arrival before unloading pigs, and the same trailers after unloading prior to departure. In total, 389 samples were collected across 26 sampling events. All samples were tested in the laboratory to detect viral genetic material. In addition, information was recorded during each visit, including environmental conditions such as temperature and humidity, trailer cleaning and disinfection practices, type of animals transported, and driver-related factors such as use of personal protective equipment (PPE), experience, and training.
The results showed that harvest facility docks are consistently highly contaminated throughout the year. Depending on the virus, between half and over seventy percent of dock samples tested positive. This indicates that the unloading area is a major hotspot for pathogen presence. A substantial proportion of trailers were already contaminated when they arrived at the plant; however, contamination levels increased further after unloading for all viruses evaluated. This demonstrates that trailers not only bring pathogens into the facility but can also acquire additional contamination during the unloading process. Importantly, 74.6% of trailers that arrive clean leave the facility contaminated with at least one virus. Seasonal patterns played a significant role in contamination risk. PDCoV and PEDV were more frequently detected during the winter months, PRRSV was more common during fall and spring, and SVA showed higher detection during summer and fall. These findings suggest that environmental conditions such as temperature and humidity may influence virus survival and transmission, and that biosecurity measures need to be implemented throughout the year. Trailer sanitation practices were identified as one of the most important controllable factors. Trailers that were washed, disinfected, and allowed to dry had significantly lower contamination rates on arrival compared to those that were not properly cleaned. In addition, trailers used to transport both pigs and cattle were more likely to be contaminated with specific viruses, suggesting that multi-species transport may increase the risk of pathogen spread across species. The study also identified important gaps in driver knowledge and biosecurity practices. Many drivers reported using PPE primarily to stay clean rather than to prevent disease transmission, and a large proportion had received little or no formal training beyond basic certification programs. This lack of awareness may contribute to inconsistent use of protective measures and increase the risk of indirect disease spread through contaminated clothing or equipment. Overall, the findings of this study demonstrate that harvest facilities are critical points for the spread of swine diseases. The interaction between contaminated docks and trailers creates a continuous cycle of contamination that can carry pathogens back to farms. For pork producers, these results emphasize the importance of assuming that trailers returning from harvest plants are contaminated and should always undergo proper cleaning, disinfection, and drying before reuse. Increasing biosecurity efforts during high-risk seasons, avoiding multi-species transport when possible, improving driver training, and minimizing risk factors during unloading are practical steps that can help reduce the spread of disease.
In conclusion, while harvest facilities represent a significant biosecurity challenge, the adoption of consistent and well-implemented sanitation practices and biosecurity measures during unloading can substantially reduce contamination risk and protect herd health.
Project #: 23-073 | Principal Investigator: Quihong Wang | Institution: The Ohio State University | Posted: 5/18/2026 | Keywords: Porcine sapovirus, vaccination, sow, diarrhea, weaning pig
Porcine sapovirus (PoSaV) infection is an emerging issue in the swine industry, affecting pigs of all ages, particularly during weaning and post-weaning, causing diarrhea and weight loss. Eight PoSaV genogroups have been identified, with GIII being the most prevalent worldwide. However, isolating circulating field strains in cell lines remains challenging. This project aimed to characterize and isolate contemporary PoSaVs from U.S. swine farms using three cell lines and to study the effectiveness of a Merck RNA particle vaccine against GIII PoSaV. For Objective 1, forty-two clinically suspicious fecal and intestinal samples were collected from diarrheic pigs in late lactation across five farms and tested for PoSaV using sensitive assays targeting viral nucleic acids. Positive samples were then serially passaged in swine testicular (ST) cells, porcine kidney (LLC-PK1) cells, and porcine intestinal epithelial (IPEC-J2) cells. PoSaV replication was confirmed by testing both viral nucleic acids and antigens. Additionally, the genomes of representative positive samples were sequenced. The overall PoSaV detection rate was 60% (25/42). Representative GIII PoSaV strains shared 87.5%-87.7% nucleotide identity in the viral capsid protein with the prototype GIII PoSaV Cowden strain detected four decades ago. The success rates for isolating field PoSaV samples were 16% (8/25) in LLC-PK1 cells, 8% (2/25) in ST cells, and 0% (0/10) in IPEC-J2 cells. Replication kinetics of an isolated strain showed earlier replication and a higher peak titer in LLC-PK1 cells than ST cells.
For Objective 2, we evaluated viral neutralizing (VN) antibody responses in the colostrum and milk samples of sows/gilts immunized with the prescribed Merck PoSaV vaccine and correlated the VN antibody titers with piglet performance. A commercial swine farm with approximately 650 sows and gilts, operating on a 4-week batch farrowing system, was selected for this study. A total of 30 animals were randomly assigned to two groups: vaccination and non-vaccination, with each group comprising 15 animals (2 gilts and 13 sows). For the vaccination group, animals were injected intramuscularly with the Merck PoSaV vaccine twice– at gestation day (GD) 73-75 (about 5 weeks pre-farrowing) and at GD 94-96 (about 2 weeks pre-farrowing). Body weight (BW) of individual piglets was obtained at birth and at 3 weeks of age. Colostrum and milk samples on parturition day, 1 week, 2 weeks, and 3 weeks post-farrowing were collected for VN antibody titers. Rectal swab samples from the sows/gilts and 3 piglets per sow/gilt at 2 weeks and 3 weeks of age were collected for monitoring PoSaV shedding. Our study found that sows and gilts within the same group had similar levels of VN antibody titers at each time point. Colostrum samples from both vaccinated and non-vaccinated groups contained high VN antibody titers (> 10^4.5 VNT50/mL), while the vaccinated group had significantly higher (p < 0.05) VN antibody titers in the colostrum and milk at 1 week post-farrowing with no significant differences at 2 and 3 weeks post-farrowing compared to non-vaccinated group. All rectal swab samples from both groups were tested negative for PoSaV, indicating no PoSaV was circulating among these pigs during this study period. Both groups of piglets had similar BW at birth and at 3 weeks of age. These results indicate that sows/gilts had been exposed to PoSaV natural infection prior to vaccination, and vaccination boosted the existing immunity. These high levels of VN antibodies in both groups of sows/gilts may provide full protection of piglets from PoSaV infection at this farm, although the threshold level of VN antibody for full protection against PoSaV has not been determined.
This study marks the first successful isolation of PoSaV strains in a new cell line (ST cells), providing a new way for future research into the viral biology and disease control and prevention (Aryal et al., 2025). We are the first to evaluate the Merck PoSaV vaccine and confirmed that it boosted existing PoSaV-specific VN antibodies in sows/gilts. However, since the pigs from both non-vaccinated and vaccinated groups did not shed PoSaV, future vaccination and challenge studies are warranted to confirm whether this vaccine is effective. (If you have any questions, please contact the PI for this project: [email protected])
Project #: 24-012 | Principal Investigator: Leonardo Cardia Caserta | Institution: Cornell University | Posted: 4/10/2026 | Keywords: NGS, sequencing, emerging viruses, metagenomics, respiratory diseases, Nanopore, rRNA depletion
Emerging infectious diseases are a constant threat to the U.S. pork industry. Early detection of new viruses is essential to control outbreaks and protect herd health. Diagnostic tests such as PCR are widely used because they are fast and accurate, but they are designed to detect only known pathogens. As a result, new viruses or highly mutated variants can remain undetected. Next-generation sequencing (NGS) can detect multiple viruses at the same time without prior knowledge of the pathogen, making it a valuable tool for identifying emerging diseases. However, its use in routine diagnostics has been limited because most genetic material in clinical samples are swine or bacteria genomes rather than viruses, which reduces sensitivity and increases costs. The goal of this project was to develop a sensitive and cost-effective NGS method for detecting viral pathogens in swine samples. We developed a procedure that removes host and bacterial ribosomal RNA from samples before sequencing, allowing viral genetic material to be more easily detected. The method was tested on 250 swine respiratory samples and detected viruses from 35 viral genera. The approach improved the detection and genome coverage of several viruses and demonstrated strong performance for DNA viruses. The estimated cost was approximately $31.54 per sample. This method can support earlier detection of emerging viruses and strengthen disease surveillance in the swine industry.
Project #: 24-030 | Principal Investigator: Leyi Wang | Institution: University of Illinois Urbana Champaign | Posted: 4/10/2026 | Keywords: PRRSV, WGS, probe capture enrichment, serum, processing fluids, tissues, oral fluids
Whole-Genome Sequencing (WGS) provides a complete “genetic fingerprint” of PRRSV, but its use has been limited by low sensitivity in standard farm samples such as oral and processing fluids. To improve the diagnostic performance of PRRSV WGS, this study evaluated a probe-capture enrichment method that acts as a “genetic magnet” to pull viral material out of complex samples. The results demonstrate a significant breakthrough: the enrichment method increased PRRSV genome coverage by nearly 46% in oral fluids, 32% in processing fluids, 25% in lung tissue samples, and 24% in serum samples, consistently capturing over 90% of the whole genome even at lower viral loads (Ct 26–27) for serum, processing fluids, and lung tissues. The improvement in PRRSV WGS means higher genome recovery when using convenient, non-invasive group samples. The validated capture enrichment-based PRRSV WGS enables faster identification of new variants, better differentiation between vaccine and wild-type strains, and more precise biosecurity decisions to protect herd health.
Project #: 24-061 | Principal Investigator: Katharine N. Bossart | Institution: Integrated Research Associates, LLC | Posted: 3/10/2026 | Keywords: Japanese encephalitis virus, diagnostic prototypes, serology, recent exposure, on-site testing
Japanese encephalitis virus (JEV) is a mosquito-borne virus that infects numerous species, including swine and humans. Although JEV is not currently in the United States, the risk of incursion remains high given the recent global expansion of JEV, which could have devastating outcomes for the swine industry, given the severe disease in piglets and pregnant sows. In the event of an outbreak, JEV diagnostics will be needed; however, there are currently no approved diagnostic tests for JEV in the United States. Additionally, the virus is classified as a biosafety level 3 agent, which significantly limits the laboratories that can work with the virus, further limiting JEV diagnostic capability. The goal of the current project was to build diagnostic capability for JEV in the United States. To accomplish this goal, JEV serology assays based on those used overseas were developed using nonhazardous recombinant JEV virus-like particles as viral antigens to broaden access and increase JEV diagnostic capability. Optimal cGMP-compliant manufacturing and purification procedures were developed for the new antigens and the new antigens were extensively characterized using a variety of biochemical and immunological assays. The newly prepared and purified antigens performed well in serological assays and performance was as good as or better than inactivated JEV; which is used as the viral antigen in JEV serological assays overseas. Two diagnostic prototype kits containing the new JEV antigens were assembled: an ELISA kit for screening numerous serum samples (requires a plate reader) and; a dot enzyme immunoassay kit for screening individual or not too many serum samples on site without advanced technology. Prototype kits are currently being evaluated overseas using ~500 swine sera from clinically confirmed cases of JEV and results should be available in the next six months. Importantly, performance data will provide real-time validation data required by regulatory agencies in the United States. Successful development of new broadly available diagnostic tests for JEV provides the swine industry with the necessary tool to detect recent JEV exposure and/or infection in swine, aiding biosecurity and control, which will help mitigate the consequences of a JEV outbreak in the United States.
It is possible that a JEV vaccination campaign would be initiated in the United States if a large scale JEV outbreak occurred. In this scenario, the ability to differentiate vaccinated and infected swine becomes important to biosecurity and control. The last objective of the current project focused on determining if a recombinant JEV nonstructural protein could be used as an alternate antigen in diagnostic kits as only infected people or animals make an immune response to nonstructural proteins. A stable cell line expressing the differential antigen was made and extensively characterized using a variety of biochemical and immunological assays. Preliminary data suggest prototype kits will not work with the alternate recombinant NS1 antigen; however, a different diagnostic serology platform was piloted which could prove useful in an outbreak.
Interest or questions pertaining to JEV prototype kits, JEV NS1 or JEV reagents can be submitted by email to [email protected] or by mail to 807 D St, San Rafael, CA, 94901.
Project #: 24-007 | Principal Investigators: Rebecca P. Wilkes | Institution: Purdue University | Posted: 2/16/2026 | Keywords: Swine respiratory disease; targeted NGS; Ion Torrent; multiplex diagnostics; pathogen surveillance
Respiratory disease continues to be one of the most costly and complex health challenges facing the U.S. swine industry. Outbreaks are often caused by multiple pathogens occurring simultaneously, making diagnosis and control difficult when relying on single-pathogen testing. While PCR remains highly sensitive, it is limited in the number of pathogens that can be tested at once and does not provide genetic information needed for surveillance and strain tracking.
This project developed and validated a targeted next-generation sequencing (tNGS) respiratory panel capable of detecting multiple viral and bacterial swine respiratory pathogens in a single test. The assay was evaluated using seventy clinical samples and demonstrated high agreement with PCR testing, while also providing additional information on pathogen strain diversity.
Results showed strong agreement between tNGS and PCR, high specificity, and robust sensitivity across clinically relevant pathogen loads. The panel successfully identified mixed infections and detected pathogens that may be missed by targeted PCR testing alone. This work supports the use of tNGS as a complementary diagnostic and surveillance tool that can improve outbreak investigation, inform vaccine strategies, and strengthen swine health monitoring programs.
Project #: 24-082 | Principal Investigators: Vlasova, Anastasia | Institution: Ohio State University | Posted: 12/15/25 | Keywords: porcine rotavirus; respiratory illness; diarrhea; suckling piglets; weaned piglets; field study
Rotaviruses, especially A and C, are among the most common causes of diarrhea in young pigs, and new evidence suggests that some strains may also affect porcine respiratory tract. Because these viruses are impossible to keep out of farms, understanding how they spread and cause disease is essential for improving herd health. Thus, the objectives of this study were: 1. To evaluate nasal and fecal rotavirus shedding by suckling and weaned piglets with three health status (respiratory signs, diarrhea, or healthy), and to determine whether RVA or RVC can be associated with respiratory disease/lesions. 2. To genetically characterize RV strains associated with respiratory disease and to define the microbiome composition associated with respiratory RV replication under field conditions.
We conducted this study in 6 swine farms in Ohio (2 research farms and 4 commercial farms). Nasal and rectal swabs were collected from suckling and weaned piglets that were healthy, experiencing diarrhea, or showing respiratory signs. Samples were tested for rotavirus RT-PCR assays. In addition, we have also analyzed tissues of 16 suckling piglets that died of undefined causes on farm 2 (research farm) to determine whether rotaviruses were present in different organs, including the organs from the respiratory tract.
Our results demonstrated that rotaviruses A and C were present on all 6 farms. While the prevalence of different rotaviruses varied greatly between different farms (A: 67-100; C: 7-56%), overall, 88% and 29% of piglets were positive for rotavirus A and C, respectively. Consistent with prior research, the highest rotavirus A prevalence/viral loads were found in diarrheic weaned piglets on most farms. However, suckling piglets with respiratory signs from farm 6 and diarrheic suckling piglets from farm 5 also had increased rotavirus A loads.
Project #: 24-043 | Principal Investigator: Marcelo Almeida | Institution: Iowa State University | Posted: 12/16/25 | Keywords:Porcine Interstitial Pneumonia, Atypical Interstitial Pneumonia (AIP), Diffuse Alveolar Damage (DAD), PRRSV, IAV, PCV2
In the last few years, the Iowa State University Veterinary Diagnostic Laboratory has seen the emergence of lesions of diffuse alveolar damage (DAD) in porcine cases. This is a histologic pattern observed in lung that is consistent with severe respiratory disease clinically. This type of lesion can be more commonly observed in cattle, but has not been observed in porcine tissues with frequency over the years. The increase in cases in recent years prompted an investigation to understand if any pathogens would be associated with it. Forty-two cases were met the selection criteria and were included in this study. PCR and IHC for PRRSV, IAV, PCV2 were performed in all cases, along with a histologic evaluation to confirm lesions of DAD and other pathogens. Next Generation Sequencing (NGS) was also performed to assess the involvement of other endemic or emergent viruses. Most cases were diagnosed with PRRSV (71.4%), IAV (35.7%), PCV2 (16.7%) or a combination of 2 or 3 of those pathogens (26.2%). PRRSV, IAV, and PCV2 were detected within lesions consistent with those pathogens in 24 (57.1%), 13 (31.0%), and 9 (21.4%) cases, respectively. However, antigen of any of those pathogens was not detected in association with lesions of DAD. NGS revealed the presence of contemporary PRRSV lineages, IAV subtypes, and PCV2 genotypes in cases associated with DAD. Seventeen viruses other than PRRSV, IAV, and PCV2 were detected; however, none of those viruses were consistently detected in all cases. Parvoviruses were detected in 45% of the cases, with parvovirus 2 and 7 detected in 21.4% and 19% of the cases, respectively. Evidence to support the involvement of any of those 17 viruses in the causation of DAD is lacking. It is unknown how PRRSV, IAV, and PCV2 infection result in this severe pattern of histologic lesions; however, control of PRRSV, IAV, and PCV2 infections through vaccination and management practices are the most likely measures to help prevent DAD in pigs.
Project #: 24-098 | Principal Investigators: Giovani Trevisan & Daniel Linhares | Institution: Iowa State University | Posted: 11/20/25 | Keywords:data analysis, surveillance, preparedness, animal health threats, diagnostic data
In 2017 SHIC funded the Domestic Swine Disease Surveillance project under the Swine Disease Reporting System (SDRS, https://fieldepi.org/sdrs/) initiative. Since then, the project has expanded significantly. This proposal aimed to maintain and enhance the SDRS program. Key objectives included: a) maintaining current PCR detection databases for major swine pathogens; b) delivering monthly PDF and audio reports to SHIC; c) updating live interactive dashboards; and d) developing a new structure for confirmed tissue diagnosis data retrieval.
From October 2024 to October 2025, twelve monthly PDF and audio reports were delivered to SHIC, summarizing diagnostic results from participating laboratories. These reports were distributed via email to 631 subscribers from 236 organizations—a 44% increase from the previous year—and reached 21 countries. Audio reports were accessed in 20 countries and distributed through podcast platforms (Spotify, Apple Podcast, Amazon Music, Google Podcast). Video summaries on LinkedIn, YouTube, and Instagram accumulated over 67,000 views.
Major updates included implementing a new PRRSV ORF5 variant classification system in the dashboards and the PRRSV BLAST tool. A new PEDV “facility” category was introduced to monitor pathogen activity in truck washes, packing plants, and vehicles. The Dx code data retrieval system was upgraded from a website query to an API-based structure, improving real-time data access and standardization.
Interactive dashboards were enhanced for usability and continue to be updated daily. SDRS remains the only publicly available source of swine health data from U.S. veterinary diagnostic laboratories, covering all age groups and specimen types. The project supports informed decision-making for veterinarians and producers. It continues supporting pathogens monitoring programs such as the PEDV Elimination Task Force and providing information for the U.S. SHIP program. International collaborations expanded with the training of two Brazilian postdocs and the developing of a Brazilian SDRS-like system (CISS). Discussions with Mexican universities may lead to similar initiatives. These partnerships enhance the U.S. swine industry’s ability to monitor global disease trends.
Project #: 24-008 | Principal Investigator: J Zimmerman | Institution: Iowa State University | Posted: 11/11/25 | Keywords: Oral fluids, Surveillance, Sampling, Sows, Group-housing
The purpose of this study was to characterize sow behaviors associated with oral fluid sampling with the intend of providing guidelines for collecting oral fluids from group-housed sows. We conducted the work on a commercial sow farm. Behavioral data were collected in 12 pens (56 of gestating sows per pen) sorted by parity (gilts, parity one, and multiparous sows) over a 4-day period. Specifically, sow oral fluid sampling behaviors were quantified by recording interactions with rope samplers over a 90-minute sampling using video cameras and then analyzing the recorded footage.
Our data showed that oral fluids can be routinely collected from group-housed gestating sows using cotton ropes – just as we already do in growing pig populations. Different from growing/finishing pigs, however, sampling time in group-housed sows should be 60 to 90 minutes in order to maximize participation. Longer sampling time is needed because fewer sows can access the rope at a time (because of their size). Further, we recommend placing 2 sampling ropes in each pen in order to provide adequate access and reduce competition for a limited “resource”. Thus, we recommend 60 to 90 minutes sampling and 2 ropes per pen in order to maximize the number of sows contributing to the sample. To reduce testing costs, the 2 samples from the same pen should be pooled prior to testing.
In addition we showed that diagnostic targets in the pen environment are transferred into pen-based oral fluid samples, just as we reported previously in finishing pigs (Tarasiuk et al. 2024. Pen-based swine oral fluid samples contain both environmental and pig-derived targets. Animals 14:766). This explains why pathogens pathogens not shed via the oral cavity (PEDV, for example) are detected in oral fluids. That is, diagnostic targets in the pen environment are picked up as pigs explore their surrounding. Subsequently, these targets are deposited in the oral fluid sample and detected by testing.
Project #: 24-093 | Principal Investigator: Dr. Michael Chetta | Institution: Talent Metrics Consulting | Posted: 10/15/25 | Keywords: safety, biosecurity, biocontainment, wean-to-market, caretaker, mitigation, prevention, preparedness, compliance, motivation, recognition
In recent years, the Swine Health Information Center has recognized that understanding what motivates farm caretakers is key to improving biosecurity. Earlier research provided support for the idea that caretakers generally want to follow biosecurity rules, but their motivation mostly came from within (personal values and beliefs) rather than from outside rewards or recognition.
The current study dug deeper into what drives caretaker behavior. Researchers focused on factors such as resources/support, feedback from supervisors, rewards, and the physical demands of the work. Caretakers in the study often pointed out that simple mistakes, inattention, or lack of action by others were the most common reasons for breaking biosecurity protocols. Many also noted that there are very few rewards or recognition for doing things right—sometimes only punishments for getting things wrong. In other words, there is little external motivation to reinforce the importance of following biosecurity procedures.
With a focus on employee motivation and attitudes, this research will aid in the creation of interventions and processes to improve resources, recognition, and effective supervision that ultimately impact how caretakers do their jobs. By learning more about the challenges caretakers face and how biosecurity is impacted, the industry can better design systems that support workers, strengthen farm practices, and further protect animal health.
Project #: 23-009 | Principal Investigator: Montserrat Torremorell | Institution: University of Minnesota | Posted: 10/15/25 | Keywords: Biosecurity, aerosols, electrostatic precipitator, air filtration, transmission, biocontainment
Costly airborne transmitted diseases in food animals are a threat to food security and the sustainability of animal farming. A commercially available electrostatic precipitator (ESP) was evaluated as an alternative to air filtration. The ESP tested in this study was highly effective at removing airborne particles with collection efficiencies similar and marginally superior to those of a MERV-16 filter. Collection efficiency was above 99% for particles above 1 µm, and for particles less than 1 µm, collection efficiency varied with temperature with higher efficiencies generally observed at lower temperatures. The ESP was also highly effective at removing PRRSV with removal efficiencies higher than 99%. The feasibility and economic analysis revealed the potential of the ESP as a biosecurity technology for sow farms, but highlighted issues on scalability, adaptation to swine farm designs, maintenance and cleaning protocols. Based on current assumptions, the ESP system had a $299,553 greater net present value over a 15-year time period, resulting in approximately $0.25 additional cost per weaned pig, when compared to air filtration. In summary, the ESP showed great performance at removing airborne particles including PRRSV from aerosols, but to be competitive its implementation in swine farms would require further development to include a design fitted for swine farms that would decrease initial and operating costs.
Project #: 23-049 | Principal Investigator: Teng Lim | Institution: University of Missouri | Posted: 10/14/25 | Keywords: Pathogen Reduction, Danish entry system, Shower in-out, Electrolyzed water, Biosecurity Protocol
The objectives of this study were to evaluate effectiveness of various biosecurity interventions, including air showers (AS), disinfectant spraying (DS), disinfectant fogging (DF), and their combinations on reducing bacterial and viral contamination on cloth, skin, and hair surfaces, using both traditional and modified protocols. Treatments were tested with and without the Danish Entry System (DES), hair nets (HN), and a modified DES (MO.DES, using hand sanitizer). Three surfaces, coverall or t-shirt, leather or pigskin (represents human skin), and faux fur (represents human hair), were contaminated with two bacteria, Staphylococcus aureus (Gram-positive) and Escherichia coli (Gram-negative), and two viruses, Canine distemper virus (CDV, enveloped) and feline calicivirus (FCV, non-enveloped), to assess the efficacy of the treatment methods. While single-step methods (AS, DS, DF) were largely ineffective on their own, integrating DES or HN significantly enhanced both bacterial and viral reductions. For bacteria (E. coli and S. aureus), DS and AS+DS achieved over 2-log reductions, with DES- or HN-enhanced combinations reaching 3–4-log reductions. While the full showering remained most effective, particularly for hair (~5-log reduction), the AS+DS+HN treatment performed similarly. Interestingly, the MO.DES eliminated bacteria to undetectable levels on hands. For viruses (FCV and CDV), only DES- or HN-based combinations achieved meaningful reductions, with AS+DS+DES and AS+DS+HN performing close to the shower protocol. Overall, DES and HN are critical additions for effective microbial control, while MO.DES and multi-step alternatives can offer practical, effective substitutes when full-body showering is impractical.
Project #: 24-017 | Principal Investigators: Giovani Trevisan and Daniel Linhares | Institution: Iowa State University | Posted: 10/2/25 | Keywords: Endemic bacteria, colibacillosis, diagnostic data, E. coli, surveillance, monitoring, SDRS
Escherichia (E.) coli is one of the most important endemic bacteria in swine health. This bacterium inhabits the pigs’ gastrointestinal tract and may be pathogenic when harboring specific virulence factors. The use of polymerase chain reaction (PCR)-based molecular diagnostics has advanced the capacity for the detection of these virulence factors. The Swine Health Information Center (SHIC) funded a project to integrate E. coli molecular detection data from major swine-centric Veterinary Diagnostic Laboratories (VDLs) participating in the Swine Disease Reporting System (SDRS). This initiative aimed to establish a centralized hub for continuous reporting of E. coli PCR genotyping results in all swine age groups, organized by individual targets, virotype (all positive targets within a sample), and pathotype, improving the level of information. Given the complex disease dynamics of E. coli, the system also allows filtering of results based on the pathogenic potential, keeping the established SDRS filters for age category and state. A user-friendly tool for veterinarians, producers, and stakeholders to monitor trends, identify risks, and make informed herd health decisions in real time was created. Results from 29,682 samples tested by PCR genotyping were collected, covering information since 2008. Samples identified as coming from research or not tagged as porcine were removed. Samples were classified as potentially pathogenic or non-pathogenic based on the virulence factors detected. Notable shifts were observed over time for fimbriae detection. The prospective data integration has been developed, and the results were first released in the monthly SDRS PDF report # 91 from September 2nd, 2025, and displayed in publicly available online dashboards with a setup for prospective daily updates (available since September 30th). From 2008 to 2017, F18 and K88(F4) had a similar detection trend, roughly being detected one or another in half of the cases. After that, F18 started being the most dominant one, peaking with 86% (911/1,062) of the detection in 2022, while K88(F4) had 10% (106/1,062) of the detection. Another important trend in potentially pathogenic samples was the hybrid ETEC/STEC pathotype detection, which increased markedly from 11% (47/419) in 2008 to 63% (514/821) in 2024, while ETEC pathotype declined from 80% (335/419) to 34% (282/821) in the same period. Overall, 30.84% (9,157/29,682) of the samples submitted to PCR genotyping tested negative for all targets. This proportion varied over time, ranging from a low of 19.4% (357/1,836) in 2023 to a peak of 40.5% (845/2,089) in 2011. The increased detection of F18 fimbriae may inform vaccine selection and be directly applied in the field. The identification of hybrid ETEC/STEC strains can further guide diagnostic decisions. Although these isolates carry Stx2e, a toxin typically associated with edema disease, the clinical presentation more frequently reported from the field was associated with enteric presentations. The negative results highlight the value of PCR genotyping, since they indicate samples that are unlikely to be associated with disease and could otherwise lead to misleading diagnostic interpretations. The E. coli PCR genotyping results were collated from major swine-focused VDLs and integrated into a user-friendly platform, where users can identify potentially pathogenic samples and generate reports by state or farm type. This platform provides valuable support to veterinarians, producers, and stakeholders. The continuous updates in the SDRS reports support the growth of the project, allowing the inclusion of additional pathogens and expanding its role as a comprehensive surveillance and monitoring tool for swine health in the US.
Project #: 24-029 | Principal Investigator: Gustavo Silva | Institution: Iowa State University | Posted: 9/18/25 | Keywords: PRRSV, spatial epidemiology, farm stratification, disease surveillance, Bayesian modeling, epidemiological modeling
This project developed a real-time, county- and farm-type–stratified spatial disease surveillance system for swine pathogens, integrating diagnostic, movement, and site location data from 3,084 sites across 18 U.S. states representing 10 major production systems. The system detects emerging diseases at the regional level and provides weekly infection risk forecasts for each production site type.
Analysis identified farm type as the primary determinant of PRRSV transmission risk (81%), followed by movement networks (16%) and geographic proximity (3%). Baseline infection probabilities were 73% for growing herds, 70% for breeding herds, and 58% for other herds. Forecast accuracy reached 83.6% for county-level models and 84.0% (95% CI: 81.2–86.7%) for site-level models. The spillover analysis (Jan 2019–Jun 2025) documented 319 breeding herd outbreaks, of which 109 were linked to potential spillover from other sites. Spillover events were characterized by state, farm type, distance between sites, and pathogen lineage. The system also detected site-specific events, enabling direct feedback to producers and supporting targeted outbreak investigations. Finally, a conditional logistic regression model assessed risk factors for PRRSV or PEDV classification in new positive sites, incorporating farm density within 5–20-mile radii around breeding herds.
This surveillance system offers data-driven, actionable insights to reduce disease spread, guide targeted interventions, and improve swine herd health at both site and regional scales. By integrating diagnostic, movement, and spatial data into a single, continuously updated platform, it enables early detection of emerging health threats, identification of high-risk sites, and real-time situational awareness for veterinarians and producers. The approach is flexible and scalable, making it adaptable to other pathogens, production systems, or geographic regions. Ultimately, this work provides the U.S. swine industry with a proactive tool to strengthen biosecurity, enhance disease preparedness, and protect animal health and productivity.
Project #: 24-016 | Principal Investigators: Giovani Trevisan & Daniel Linhares | Institution: Iowa State University | Posted: 9/10/25 | Keywords: Diagnosis, surveillance, endemic diseases, animal health threats, diagnostic data
The Swine Health Information Center (SHIC) funded a project to generate a data-driven disease index to monitor swine pathogens’ activity in the U.S. using confirmed tissue-based diagnoses from the Iowa State University Veterinary Diagnostic Laboratory (ISU-VDL). This initiative aimed to provide a transparent, automated, reproducible method to help veterinarians, producers, and stakeholders prioritize disease threats based on real-world diagnostic data.
The index was built using 59,950 porcine cases from 2020 to 2024. It considered four key factors: how often a disease was diagnosed, how often it appeared alongside other diseases, how widespread it was across U.S. states, and how frequently it triggered statistical alarms for unusual activity within a year. These factors were weighted and combined into a single score for each disease, updated weekly for the ongoing year, and displayed in an interactive Power BI dashboard. The results confirmed that PRRSV and Streptococcus suis remain the top two-ranked pathogens, demonstrating their high activity in the U.S. swine industry. The system also detected emerging pathogens’ activity in 2024, including porcine sapovirus and astrovirus, while PCV2 showed a notable decline. The dashboard allows users to track disease trends and compare rankings year-to-year, supporting decision-making. The index was validated using Euclidean and Manhattan distance models to assess year-over-year consistency and detect emerging or declining disease trends. Bootstrap resampling (500 iterations) generated 95% confidence intervals for index predictions, excluding EARS due to model limitations. The index was integrated into a Power BI dashboard for real-time visualization and weekly updates.
The disease index monitors swine pathogen activity, identifying emerging threats. The index creates possibilities for being adapted and integrated into the Swine Disease Reporting System (SDRS) and expands its use across other livestock sectors.
Project #: 24-003 | Principal Investigator: Erin Kettelkamp | Institution: Swine Vet Center | Posted: 8/14/25 | Keywords: Biosecurity, Decontamination, Disinfection, PEDV, Swine, Trailer
Objective
The overarching aim of this study was to evaluate the effectiveness of a waterless trailer decontamination method using modified vaporous hydrogen peroxide (mVHP) in combination with an industrial vacuum system on PEDV detection and inactivation. To do so, the effect of mVHP on detectable PEDV RNA levels using a waterless swine trailer decontamination system and varying disinfectant holding times was evaluated. In addition, the antiviral efficacy of mVHP against PEDV was assessed using a bioassay.
Research Conducted
An experimental study was designed to understand the effect of PEDV detection and inactivation using an mVHP system and bioassay challenge model. For this study, two scenarios were considered: 1) Mock-swine trailer under in-vitro conditions (i.e., chamber) and 2) Mock-swine trailer under field-simulated conditions (i.e., shroud). For the second scenario, a miniature insulated trailer “shroud” was designed to mimic conditions for potential field application. A miniature aluminum trailer model was contaminated with PEDV fecal inoculum in each scenario. An industrial-grade vacuum was used to remove organic material, followed by applying mVHP treatment with differing holding times. Pre-marked trailer surfaces were sampled before and after treatment to confirm PEDV contamination and to assess differences in PEDV RNA detection. A bioassay was performed to determine the role of mVHP treatment and differing holding times on PEDV infectivity, utilizing liquid that was recovered from the trailer post-treatment.
Research Findings
Applying mVHP following vacuum removal of bulk material reduced detectable levels of PEDV RNA from contaminated trailer surfaces compared to untreated controls. No differences in PEDV detection levels via Ct values were observed based on holding time. In addition, numerically similar results were observed between the chamber and shroud scenarios. The antiviral efficacy of mVHP treatment against PEDV could not be determined, as all extracted trailer samples failed to produce PEDV infection under in-vivo conditions. All pigs remained clinically healthy during the bioassay and tested negative for PEDV using RT-PCR. In addition, the absence of PEDV infection via IHC and intestinal scoring was shown. Under the conditions of this study, mVHP treatment paired with a vacuum system reduced the relative PEDV load (i.e., Ct value) from contaminated trailers. Further work is needed to assess the impact of this technology on virus inactivation and its potential application and scalability in the field.
Implications for the Industry
Combining an industrial vacuum and mVHP treatment offers a promising alternative to traditional trailer sanitation methods. Unlike current wash and TADD procedures, this system is portable, waterless, and scalable, thus reducing labor, infrastructure, and water requirements associated with existing practices. This technology may be particularly valuable during a disease outbreak when the rapid and thorough decontamination of swine transport vehicles is critical. In this pilot study, mVHP treatment of contaminated trailers showed a reduction in the levels of detectable PEDV genetic material via RT-PCR. The role of mVHP treatment on PEDV infectivity was inconclusive; therefore, additional research is needed to validate virus inactivation capabilities and to improve the standardization of contamination methods to optimize bioassay procedures. With continued development, this approach could play an important role in improving transport biosecurity and disease outbreak response readiness in the swine industry.
Project #: 25-053 | Principal Investigator: Dr Brendan Cowled | Institution: Ausvet Pty Ltd | Posted: 6/30/25 | Keywords: Australia, Japanese encephalitis virus, Epidemiology, Outbreak, US Preparedness, US Response
Ausvet Contact Information
Brendan David Cowled ([email protected], +61 4 20851350)
Background
Japanese encephalitis virus (JEV), a mosquito-borne virus not found in the US, significantly impacts domestic swine, causing severe reproductive failure. Australia experienced a JEV outbreak in its swine industry in 2021-2022.
Objectives of Research
Our objectives were to understand how and why JEV spread in pigs and to make recommendations to assist the US industry in preparing in case JEV should ever arrive in the US.
How did we do this research?
We employed two methods:
Interviews:
We interviewed Australian swine veterinarians and experts for their perspectives on the JEV outbreak.
Data Analysis:
We analyzed industry production, environmental, and meteorological data to identify drivers of JEV spread.
Results and key producer outcomes
What was seen on infected swine farms in Australia
JEV effects on swine farms varied, from 50-60% of affected litters to minimal or no apparent disease. Common observations included mummified fetuses, reduced conception rates, smaller litters, stillborn or weak “shaker” piglets, and prolonged gestation. Boars frequently experienced infertility and inflamed testicles. While initial surges in affected litters often declined rapidly (4-6 weeks), some herds saw impacts over 10-12 weeks.
What was causing disease and control
Increased rainfall contributed to the 2021-2022 outbreak. It’s believed wild waterbirds spread JEV down Eastern Australia, interacting with increased mosquito populations, leading to a transmission cycle that spilled over to domestic pigs. Controlling JEV across the general landscape (wild waterbirds and mosquitoes) was impossible.
Control efforts primarily focused on interventions at the individual swine farm level, alongside public health measures like human vaccination and mosquito bite prevention. For swine, without a vaccine, mosquito control was key. This included enhanced monitoring, environmental and chemical control (removing water sources, larvicides, residual insecticides), and direct treatments (e.g. insecticidal pour-ons). Longer-term measures included vaccine trials and development. Mechanically ventilated systems were more effective at excluding mosquitoes.
Risk Factors
A complex relationship between rainfall and land type around farms influenced JEV risk. Wetlands, subtropical grasslands, and increased rainfall correlated with higher disease incidence. Conversely, more temperate grasslands provided protection, even with heavier rain. These risk factors can inform AI-driven predictions for high-risk periods, guiding targeted control and surveillance.
Recommendations and key producer outcomes
We have proposed a comprehensive suite of recommendations in our full reports. These are divided into preparation for a possible outbreak, response to a confirmed outbreak and what to do if JEV becomes established in the US.
Summary and conclusion
As JEV’s global range expands due to changing weather and migratory patterns, Australia’s experience offers crucial lessons for commercial swine industries like the US. Our qualitative and quantitative research identified key lessons to help the US swine industry respond and adapt to a potential JEV outbreak.to JEV.
Project #: 23-077 | Principal Investigator: Michael C. Rahe | Institution: North Carolina State University | Posted: 6/23/25 | Keywords: PoAstV4, tracheitis, bronchitis, CDCD pigs, machine learning, immunohistochemistry, in situ hybridization
Porcine astrovirus 4 (PoAstV4) has previously been detected in piglets with respiratory disease. However, it was unclear if the virus was the cause of disease or merely a bystander. The objective of this study was to infect piglets with PoAstV4 and evaluate if respiratory disease was reproduced. Caesarean-derived colostrum-deprived (CDCD) piglets were infected (17 – challenged and 11 negative controls) with PoAstV4 and were found to be shedding the virus in nasal secretions as early as 2 days post challenge (DPC) with all pigs testing negative by 14 DPC. Both tracheitis and bronchitis were diagnosed in infected pigs necropsied at 5 and 8 DPC with viral detection in these tissues at the same time. There was a productive immune response to infection with detection of anti-PoAstV4 antibodies in the blood and the characterization of immune cells within infected tissues. Results from this study show that PoAstV4 can cause microscopic lesions of an epitheliotropic viral infection in the respiratory tract, very similar to influenza A virus.
Project #: 23-068 | Principal Investigator: Igor Paploski and Cesar Corzo | Institution: University of Minnesota | Posted: 4/11/25 | Keywords: epidemiology, tongue tips, disease testing, surveillance, PRRSV, swine, infectious diseases
Porcine reproductive and respiratory syndrome virus (PRRSV) causes significant economic losses in the U.S., approximately USD 1.2 billion annually, due to reproductive failure, abortion, and high pre-weaning mortality among piglets. Approximately 30% of the U.S. breeding herd experiences a PRRSV outbreak every year. Tongue tips from dead animals, particularly piglets, are being considered as an alternative specimen to monitor PRRS during herd stabilization; however, questions as to how to better process these samples to optimize the sensitivity when detecting disease still exist. This study aimed to describe the impact of different tongue tips processing and testing protocols to optimize the sensitivity and specificity of PRRSV detection in sow herds. Samples from seven farms were tested using different pooling strategies, processing techniques, and storage conditions. Results showed that testing tongue tip fluids yielded more sensitive PRRSV detection compared to tongue tissue homogenates, and that keeping samples frozen yield lower Ct values when compared to refrigerated samples. Pooling samples reduced diagnostic accuracy but can still provide valuable information depending on the question being addressed. We suggest that practitioners discuss testing objectives with pathologists before sample submission. We also estimate that for every day elapsed since tongue tip collection, the Ct value increase by 0.2 units, suggesting that delays in sending the samples and shipment should be avoided. Tongue tips are an easy-to-collect sample type that targets animals potentially more likely to be infected (dead piglets), diminishing welfare concerns during sample collection. This study provides valuable insights into how testing choices and submission circumstances impact RT-PCR PRRSV testing results of tongue tips.
Project #: 23-078 | Principal Investigator: Cesar A. Corzo | Institution: University of Minnesota | Posted: 3/13/2025 | Keywords: Monitoring, Swine Health, PRRS, PEDv, Senecavirus, SVA, PRRSv 1H.18, PEDv Stability
Objective 1: Monitor trends in pathogens incidence and prevalence – PRRSv, PEDv, PDCoV, Senecavirus and central nervous system associated viruses continued to be monitored, each maintaining historical patterns. During 2023-2024 we explored and developed a method to estimate the breeding herd Senecavirus cumulative incidence. Fortunately, cumulative incidence remained below 2.5%, with most of the years remaining below 0.5%. Another objective was to estimate the time herds required to eliminate PEDV and explored associated factors. A significant reduction in time to consistently wean RT-PCR negative piglet was observed when comparing epidemic (i.e., 24 weeks) versus endemic (i.e., 13 weeks) stages of the disease in the US. Factors such as previous immunity, herd size, season when outbreak occurred were associated with the time to wean negative PCR piglets.
Objective 2: To conduct prospective monitoring of PRRSv sequence evolution and impact – During 2023-2024 we continued to curate our PRRSv ORF5 database. The representativeness of this database has enabled multiple collaborations, including outbreak investigations and, most recently, the development of the new PRRSv classification system. Thanks to this new classification we were able to identify a new variant of concern allowing us to communicate to the industry of this finding in a timely manner. Fortunately, this variant does not seem to have the transmissibility that the L1C.5 had; however, vigilance remains necessary. Lastly, we developed a mechanism to identify herds that are having a prolonged time-to-stability and are currently beta testing this methodology.
Objective 3: To expand participation of producers to allow for all to be involved – During 2023-2024 we added one production system and are awaiting enrollment forms from a second system. In addition, our website continued to be finetuned and updated with the information our readers are requesting, and we have reached over 15,000 views. The most visited section is related to reports, totaling 1,400 views. Interestingly, we are seeing that MSHMP website visitors are located around the world, but both the US and China are the main consumers. During 2024, our team and collaborators published a total of nine peer reviewed manuscript which speaks for the relevance and versatility of the dataset.
Project #: 23-029 | Principal Investigator: Gustavo Silva | Institution: Iowa State University | Posted: 3/11/2025 | Keywords: Biosecurity, wean-to-harvest, PRRSV, PEDV, swine
Objectives: The main objectives of this proposal were: 1) to assess the current bioexclusion practices used at wean-to-harvest sites across the U.S., ensuring a diverse group of producers from different swine-producing states are included; and 2) develop a tool that veterinarians, production managers, and producers can use to assess biosecurity on their sites quickly.
Research methodology: This study assessed biosecurity practices at wean-to-harvest sites across major U.S. pork-producing states. Data were collected through a questionnaire completed by 21 herd veterinarians, including production systems and independent producers. The questionnaire was developed with input from industry experts and comprised 69 questions on bioexclusion practices, covering site characteristics, vehicle movements, people movement, manure removal, water entry, and sanitation. A weighted method ensured the results reflected all respondents’ answers. For the second phase, we enrolled 139 wean-to-harvest sites to assess biosecurity practices and their relationship with lateral disease introduction of PRRS, PEDV, PDCoV, and TGEV. Farms must be stable or negative for PRRS and key enteric viruses, including PEDV, PDCoV, and TGEV. Participating producers were asked to complete a biosecurity questionnaire with 115 questions covering risk events, biosecurity management practices, herd demographics, trucking sanitation, and farm location.
Research findings: the results of phase 1 include data from 15.7 million pigs across 3,680 sites in 13 states. Of the 3,680 sites, 10.3% were nurseries, finishing represented 52.9%, and 36.8% were wean-to-finish sites. 93.3% of the farms reported using all-in-all-out, mortality disposal was mostly off-site (65.3%), and 47.3% of employees visited more than one site daily. While most sites have shower facilities (63.8%), fewer require employees to shower in (57.6%) or out (56.9%). Manure is removed about 1.5 times yearly, often by third-party companies. Most sites rely on well water (87.7%), but some don’t perform any water treatment (64.7%). Trucks hauling pigs are generally washed and disinfected, with 100% of trucks hauling weaned pigs cleaned between loads. For feeder trucks, 60.9% are washed, and 63.9% are disinfected between every load, and for market hog trucks, 78.3% are washed, and 52% are disinfected between every load. For the second phase, the data of 139 sites across nine companies in six states enhanced 44 nurseries from three companies, 44 finishers from three companies, and 51 grow-finish sites. The data shows that PRRSV outbreak rates were highest in grow-finish sites (61.4% – 27/44), followed by wean-to-finish (52.9% – 27/51) and nurseries (34.1% – 15/44). No outbreaks of PEDV or coronaviruses were reported in nurseries or wean-to-finish sites but grow-finish sites had a break rate for coronaviruses (2.3%) and a higher rate for PEDV (11.4%). Key findings include that nursery sites had 92% lower odds of reporting a PRRSV outbreak than finishers, and biosecurity practices like bench entry, truck washing, and downtime between loads reduced outbreak risk. Hauling animals with unknown status for PRRSV increased the odds of reporting an outbreak by 12 times, stressing the need for careful animal health monitoring before transportation.
Industry implications: The study’s first phase highlighted that while most sites reported implementing biosecurity measures like vehicle washing and employee training, gaps still need to be addressed, especially in communication and compliance auditing. The second phase revealed that nursery sites have a significantly lower risk of PRRSV outbreaks than grow-finish sites, which face a much higher risk. This emphasizes the need for stronger biosecurity in the finisher phase. Simple, cost-effective measures like bench entry—where employees change footwear or clothing before entering different areas—can help reduce the spread of PRRSV and are easy to implement. While these early findings are promising, more data is needed to refine biosecurity recommendations and help producers improve their practices, enhance surveillance, and build a more resilient industry.
Project #: 23-063 | Principal Investigator: Cesar A. Corzo | Institution: University of Minnesota | Posted: 2/19/25 | Keywords: Post-mortem, tongue tip, growing pigs, alternative specimens
In the US swine industry, most post-weaning health monitoring sampling relies on oral fluid sample collection since jugular venipuncture can be time-consuming and requires skilled personnel. Assessing whether easy-to-collect post-mortem samples, can provide value for diagnosis is needed as labor constrains are a concern in today’s industry. Furthermore, finding practical and time-efficient methodologies to monitor health during the post-weaning stages is necessary as this methodology can be rapidly adopted by the industry leading to a better understanding of disease dynamics. Recently, European researchers reported for the first time on the use of tongue tip fluids (TTF) sampling to detect PRRS given that their laws prohibit them from castrating piglets, thus, processing fluids was not an option for monitoring. In the US, work on TTF has been in preweaning animals but data on post weaning pigs is virtually non-existent. Therefore, the objectives of this study were 1) to assess the sensitivity and specificity of TTF, and other specimens including intracardiac blood (IC), oral/nasal swabs (ONS), rectal swabs (RS) and superficial inguinal lymph nodes (SILN) in growing pigs for the detection of PRRSV; and, 2) to characterize the detection of Porcine Circovirus type 2 and 3 (PCV2, PCV3), Porcine Parvovirus type 1 and 2 (PPV1, PPV2), Lawsonia intracellularis (Li), and Influenza A virus (IAV) in TTF, TTF, ONS, RS, and SILN.
Two growing pig farms located in Minnesota and representative of current swine production practices were included in this study. The first farm, a 2,400-head wean-to-finish farm undergoing a PRRS outbreak was visited when pigs were 6 and 12 weeks of age (WOA). The second farm was a 3,300-head finishing site undergoing a similar health challenge as farm 1 and was visited when pigs were 15 WOA. During each farm visit, a total of 30 dead pigs were sampled, resulting in a total sample of 90 pigs. From each pig, TTF, IC, ONS, RS and SILN samples were collected and tested in a way that for specific pathogens a gold standard sample was selected and then compare it with TTF results. Briefly, all TTF samples were tested individually by RT-PCR for all pathogens (i.e., PRRSV, SIV, PCV2/3, PPV1/2 and Li). All specimens were tested individually for PRRSV, while additionally ONS was individually tested for IAV; RS for PPV1, PPV2, and Li; while SILN were individually tested for PCV2 and PCV3. The sensitivity (Se), specificity (Sp), positive predictive value (PPV), and negative predictive value (NPV) were calculated for PRRSV TTF and IC serum as the gold standard. The proportion of RT-PCR positive results from specimens tested for other pathogens was compared by descriptive statistics. Most pathogens were detected at least once in TTF with Ct values ranging from 11.6 to 39.8. For PPRSV the best results for all specimens were at 11 WOA, when TTF had Se=84%, Sp=9%, PPV=62%, NPV=25%, ONS had Se=74%, Sp=73%, PPV=82%, NPV=62%, and SILN had Se=100%, Sp=9%, PPV=66%, NPV=100%. PCV2 was detected in 43% of TTF and 11% of SILN samples, PCV3 was not detected in any sample. PPV1 was detected in 1% of TTF and 0% of RS, PPV2 was detected in 97% of TTF and 61% of the RS samples. Li was detected in 6% of TTF and 0% of the RS samples. IAV was detected in 38% of TTF and 38% of the ONS samples.
Most pathogens were detected on TTF samples during the three different ages indicating that this specimen can provide valuable post-mortem information during a diagnostic investigation. Detection of some pathogens in TTF could be the result of shedding or contamination which should encourage practitioners and veterinarians to interpret results with caution when using TTF. In our case, the overall diagnostic performance of all the other specimens used besides TTF still requires further investigation. Indeed, a complete and exhaustive collection of multiple clinical specimens from different body systems remains the standard in diagnostic investigations.
Contact: [email protected]
Project #: 23-052 | Principal Investigator: John J McGlone | Institution: Texas Tech University | Posted: 2/19/25 | Keywords: Pigs, Vaccination, Environmental Enrichment, Erysipelas, Ileitis
Environmental enrichment (EE) devices or programs are required in some countries and in some markets. Any EE device that has a second purpose would more likely encourage adoption. We developed an EE device that allows pigs to self-administer liquids by building the EE device consistent with pig rooting, investigating and play behaviors. A pilot study demonstrated pig preference for the EE device when a maternal pheromone was sprayed on the device. We previously have shown that this method of vaccine delivery was efficacious for pigs to self-deliver a Salmonella vaccine. In this study, we sought to determine if the EE self-administration device could deliver vaccines for four diseases common among growing pigs. A baseline sample determined the antibody status of subjects. Assay for serum IgG and IgA determined efficacy with three treatments groups: (1) a control that was not vaccinated, (2) a group in which pigs were individually vaccinated by oral gavage or intramuscular (i.m.) injection, and (3) a self-vaccinated group. Self-vaccination was efficacious for Erysipelas and Ileitis vaccines in that pigs built robust titers to these antigens. Self-vaccination using commercially licensed vaccines at labeled doses and timing for Influenza and Mycoplasma hyopneumoniae did not stimulate serum or oral fluid antibodies. Using EE for self-vaccination of selected vaccines is possible today which will reduce labor needs, eliminate the need for needles, and will allow pen-level vaccinations or delivery of other animal health products (drugs, pheromones, etc.).
Project #: 22-003 | Principal Investigator: Yi Lu | Institution: University of Texas at Austin | Posted: 2/14/25 | Keywords: aptamers, PRRSV, PEDV, sensors, detection
Swine viral pathogens, such as Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) and Porcine Epidemic Diarrhea Virus (PEDV), pose significant economic challenges to the global swine industry. To ensure prompt and effective management of these swine viral outbreaks, novel diagnostic tools that allow on-site and real-time detection are required. While diagnostic methods have been developed and are available, they either require sample pretreatment and a skilled operator performed on a costly equipment that takes hours to days in a professional laboratory and thus not suitable for on-site detection, or they cannot tell whether the virus in infectious or not, causing delays in managing the viral outbreak.
To meet these challenges, we have developed a novel method for direct detection of intact viruses without any sample pretreatment, with the ability to detect and differentiate infectious swine viruses such as PRRSV and PEDV from the same virus that has been rendered noninfectious by disinfection. The method is based on DNA aptamers that can be selected to bind and differentiate infectious swine viruses from noninfectious and other viruses. By immobilizing the aptamers into a nanopore, only infectious PRRSV or PEDV will produce electrochemical signal changes and thus can be detected and quantified using a handheld meter.
Our primary objectives for this project are as follows:
1. To obtain DNA aptamers that can bind infectious PRRSV and PEDV through in vitro selection and counter selection processes, with the aim of enhancing selectivity.
2. To design and validate DNA aptamer-nanopore sensors for the direct detection of infectious PRRSV and PEDV, both within controlled cell cultures and real-world field samples.
To achieve the objectives of this project, we conducted ten SELEX (Systematic Evolution of Ligands by Exponential Enrichment) experiments under varying conditions to optimize aptamer selection for porcine reproductive and respiratory syndrome virus (PRRSV). Through this process, we identified virus purity as critical to ensuring efficient aptamer selection throughout SELEX. Sequencing of SELEX DNA pools, followed by bioinformatic analysis, enabled us to identify promising aptamer candidates targeting PRRSV. Biochemical characterization of these candidates demonstrated that several could selectively bind to infectious PRRSV II, with minimal binding to noninfectious PRRSV II. While promising in vitro, additional optimization or complementary techniques may be required for field applications. The insights gained from this study underscore the importance of sample purity and the need to further enhance aptamer selectivity to improve detection reliability in applied settings.
Contact info for PI of the project: [email protected]
Project #: 22-059 | Principal Investigator: Gustavo Machado | Institution: North Carolina State University | Posted: 1/1/25 | Keywords: Truck, transport, disease modeling, contact trace, indirect contact, truck cleaning, and disinfection.
Jason A. Galvis1 and Gustavo Machado1
1Department of Population Health and Pathobiology, College of Veterinary Medicine, North Carolina State University, Raleigh, NC, USA.
Introduction: Disease transmission via farm-to-farm transportation vehicles is still to be fully appreciated as a significant route of disease dissemination; however, it has slowly sounded the alarm for swine and systems. While transmission by vehicle movements has been associated with outbreaks such as foot and mouth disease (FMD) and African swine fever (ASF), it is still unknown how these movements represent a risk for disease transmission. This is due to the absence of information about pathogen viability on vehicle surfaces and the efficacy of cleaning and disinfection to eliminate such pathogens.
Methodology: In this study, we developed a novel methodology to reproduce the indirect contact network among farms by vehicle movements, which combines pathogen stability, conditioned to decay over time, with environmental temperatures. In addition, we included that pathogens may be eliminated by an effective cleaning and disinfection event. Because we did not know the actual efficacy of cleaning and disinfection procedures, we simulated six different cleaning efficacies: 0, 10%, 50%, 80%, 90%, and 100%, which are triggered each time a vehicle contacts a cleaning station. To ensure vehicles are a source of infection due to their proximity to the farms, we identified farm locations using the Perimeter Buffer Area (PBA) collected from RABappTM. Thus, a vehicle contacted a farm if it came within a determined distance from the PBA. To test this methodology, we collected movements from 567 vehicles across three swine companies in two regions of the U.S. These vehicles were divided into six transportation types depending on if they transported people, pigs to farms, pigs to market, feed, undefined, and one additional type combining all vehicles. For each vehicle type, we reconstructed a contact network and evaluated the median number of farms that could be infected by considering the chronological order of contact for one year.
Results: In region one, after a year of movements and without effective cleaning (0%), the combined vehicles were able to potentially infect a maximum of 2,157 farms, similar to the results of vehicles transporting feed (Figure 1). This result was lower for vehicles transporting pigs to farms (2,089), pigs to market (1,507), undefined vehicles (1,760), and personnel (3). However, with 100% cleaning efficacy, the farms in the contact chain of pooled vehicles, vehicles transporting feed, and undefined vehicles was reduced by 1%. This reduction continued with 26% for vehicles transporting pigs to farms, 43% for vehicles transporting pigs to market, and 66% for vehicles transporting crew. In region two, which only had one vehicle transportation role with an inefficient cleaning efficacy (0%), undefined vehicles can potentially infect 437 farms, and this number decreased by 76% with 100% cleaning efficacy.
Discussion and conclusion: Except for vehicles transporting crew, all other vehicle types exhibited the potential to disseminate disease to numerous farms in both regions. For vehicles
transporting feed and undefined vehicles in region one, increased cleaning efficacy showed a low impact in reducing potentially infected farms, which is associated with a low frequency of cleaning events. On the contrary, for vehicles transporting pigs to farms, pigs to market, and undefined vehicles in region two, an increased cleaning efficacy showed a higher reduction of potentially infected farms due to a higher frequency of which trucks drove through cleaning stations. This is likely related to a higher perception of risk due to these vehicles having direct contact with organic material by transporting live animals. In conclusion, even when vehicle cleaning and disinfection is fixed at 100% efficacy, vehicles can create numerous paths in the network to spread swine diseases. Thus, new strategies are necessary to reduce the risk of transmission by vehicle movement, such as redirecting routes or increasing the frequency of cleaning.
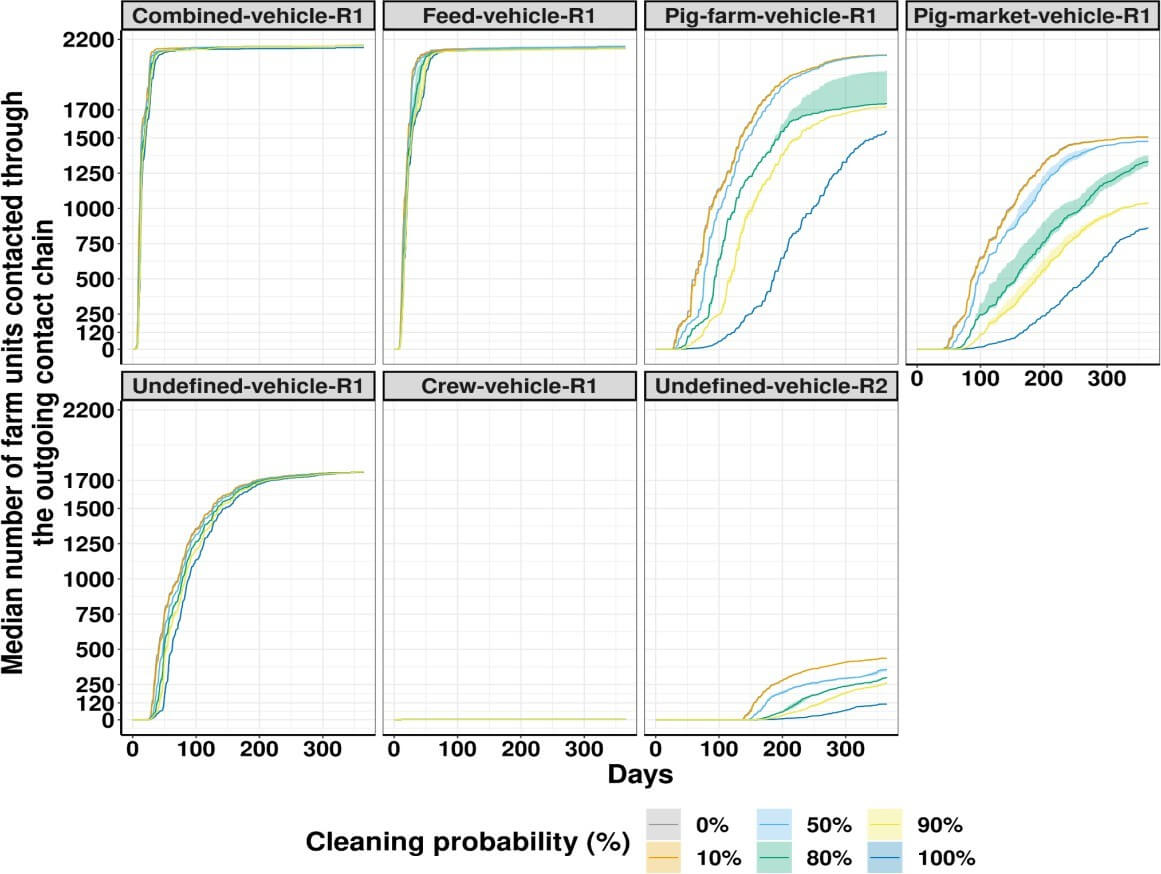
Figure 1. The number of farm units potentially infected via vehicle movements. Solid lines represent the median, while shadowed areas represent the interquartile ranges from ten simulations performed for each cleaning probability. Region one is identified by R1 and region two by R2. Combined-vehicles represent the combination of all vehicles movements from region one, feed- vehicle are vehicles transporting feed to farms, pig-farm-vehicle are vehicles transporting live pigs to farms, pig-market-vehicle are vehicles transporting live pigs to market, crew-vehicle vehicles are vehicles transporting farm personnel, and undefined-vehicles are vehicles without a defined role.
Project #: 23-076 | Principal Investigators: Isadora Machado, DVM MSc., and Daniel Linhares, DVM MBA PhD. | Institution: Iowa State University | Posted: 12/13/24 | Keywords: PRRSV, tongue fluids, risk-based, monitoring, targeted sampling, swine
Objectives: The primary objective of this study was to evaluate tongue fluids (TF) as a risk-based sampling approach in commercial breeding herds. TF PRRSV reverse transcription-qualitative polymerase chain reaction (RT-qPCR) results from stillborn piglets were evaluated as an indicator of PRRSV RNA detection in liveborn littermates. Secondly, we evaluated the presence of stillborns and litter size as indicators of PRRSV detection in live piglets.
Methodology: Samples were collected from two PRRSV-positive breeding herds (Herd A: 2,500-sow farm with a three-week batch farrowing system; Herd B: 4,500-sow farm with a weekly batch farrowing system). A total of 130 litters were sampled within 12 hours after farrowing: 66 without stillborn piglets and 64 with stillborn piglets, totaling 1,723 liveborn and 105 stillborn sampled piglets. Regarding the litter’s parity: in Herd A, 14 were parity 1 sows, 6 were parity 2, 4 were parity 3, and 1 parity 4; in Herd B, all 104 litters came from parity 1 sows.
TF and intracardiac blood (IB) samples were individually collected from stillborn piglets, and tail blood swabs (BS) were individually collected from all liveborn littermates within the selected litters. Samples were individually tested for PRRSV RNA detection by RT-qPCR at the Iowa State University Veterinary Diagnostic Laboratory. Litters with ≤ 11 liveborn piglets were defined as small litters, whereas litters with ≥ 12 were defined as large. Generalized linear regression models were used to evaluate:
– Incidence of stillborn piglets on the probability of PRRSV RNA detection within the litter;
– Stillborn TF PCR-result as an indicator of PRRSV in liveborn piglets;
– Litter size as a risk factor for PRRSV.
Research Findings: Considering all sample types (TF, IB, and/or BS), the percentage of PRRSV-positive litters based on PRRSV RNA testing was 21.5% (28 of 130 litters), of which 15.4% (20 of 130 litters) were BS PRRSV RNA-positive, 10% (13 of 130 litters) were IB PRRSV RNA-positive, and 16.9% (22 of 130 litters) were TF PRRSV RNA-positive. At an animal-level, considering both herds, 3.4% (59 of 1,723 liveborn piglets) were BS PRRSV-RNA positive, 16.2% (17 of 105 stillborn piglets) were IB PRRSV-RNA positive, and 26.7% (28 of 105 stillborn piglets) were TF PRRSV-RNA positive (Table 1). The mean positivity of liveborn piglets within the litter was 5%, varying from 0% to 85.7%, while the total born (stillborn and liveborn piglets combined) was 4.6% (0% to 76.4%).
Regarding sample types’ Ct values, BS had a median of 35.5 (range from 19.3 to 39.9), IB a mean of 22.4 (range from 16.1 to 36.7), and TF a mean of 29.2 (range from 24.6 to 38.5) (Figure 1). Intracardiac blood exhibited the lowest median Ct value, which was significantly different from the mean Ct values of the sample types (P-value < 0.05).
In litters with at least one stillborn compared to those without a stillborn, the odds of having a PRRSV RNA-positive result (IB, TF, or BS) increased 5.6 times (95% confidence interval [CI]: 2.21–16.28, P-value < 0.001), and the odds of having at least one viremic liveborn littermate (BS) increased 2.8 times (95% CI: 1.04–8.40, P-value = 0.049). The probability of detecting PRRSV in litters with stillborn was 35.9% (25.2%, 48.3%), compared to 9.1% (4.1%, 18.8%) in large litters. When the stillborn TF was positive, the odds of having a viremic liveborn littermate (BS) increased 20.8 times (95% CI: 6.90–69.56, P-value < 0.001).
Small litters had 5.3 times higher odds (95% CI: 2.21–13.13, P-value = 0.002) of yielding a positive result (IB, TF, or BS) than large litters. The probability of detecting PRRSV in small litters was 45.7%, compared to 13.7% in large litters. In small litters, the odds were 7.4 times higher (95 % CI: 2.72–21.91, P-value < 0.001) for having at least one positive liveborn piglet and 2.7 times higher (95 % CI: 1.05–7.19, P-value = 0.03) for having at least one positive stillborn piglet. Results are summarized in Table 2. Lastly, litter size using total born per litter had no confounding effect and was not statistically significant; therefore, it was not included in the final model. Results are summarized in Table 2.
Industry Implications: Litters with stillborn piglets and small litter size had a higher probability of being PRRSV RNA-positive. Moreover, TF samples from stillborn piglets are a reliable indicator of the PRRSV status of their liveborn littermates, indicating that TF can be an effective risk-based approach for PRRSV detection. Therefore, veterinarians and pig producers are encouraged to collect TF, targeting stillborn piglets and small litters to increase the likelihood of detecting PRRSV. This risk-based sampling strategy within the first hours post-farrowing enhances the effectiveness of PRRSV monitoring programs in breeding herds that support timely interventions, such as McREBEL and cross-fostering. Lastly, while TFs were individually collected in this study to test the hypothesis, collecting them as an aggregated sample, similar to processing fluids, is recommended. This approach increases the likelihood of detecting the virus, allowing for a larger number of animals to be screened.
Project #: 23-053 | Principal Investigator: Nubia Resende de Macedo | Institution: Iowa State University | Posted: 12/12/24 | Keywords: Glaesserella australis, Diagnostics, Swine, Novel, Pathogen
Industry Summary: Glaesserella australis, a newly recognized Gram-negative bacterium species, was isolated from the lung lesions of pigs in Australia in 2018. More recently, in 2023, G. australis was also identified in two swine herds in Ontario, Canada. The isolates were obtained from the pericardium and lung tissues of pigs. The hypothesis remains that G. australis has been misdiagnosed over the years since the disease presentation is similar to porcine pleuropneumonia caused by Actinobacillus pleuropneumoniae, and no methods were being used to detect G. australis accurately.
Sequencing of historical isolates with similar characteristics was attempted at the ISU VDL, but no match was found. Therefore, the G. australis reference strain was obtained and added to our sequencing platform and, most importantly, to our MALDI-TOF database for real-time screening of clinical samples. Additionally, an RT-PCR is now available for further screening and diagnostics purposes. An ISH assay is also being developed to detect this organism in tissues associated with lesions.
At the ISU VDL, since January of 2024, we have screened for G. australis through MALDI-TOF, but this new potential swine pathogen is yet to be found. Nevertheless, the tools described here will contribute to the G. australis screening process in the U.S. and help improve our understanding of its prevalence and pathogenesis.
Project #: 23-028 | Principal Investigator: Gustavo Machado | Institution: North Carolina State University | Posted: 1/10/25 | Keywords: Trucks, deliveries, transportation, prevention strategies, and biosecurity
Jason A. Galvis1, Cesar Corzo2 and Gustavo Machado1
1Department of Population Health and Pathobiology, College of Veterinary Medicine, North Carolina State University, Raleigh, NC, USA.
2Veterinary Population Medicine Department, College of Veterinary Medicine, University of Minnesota, St Paul, MN, USA
Introduction: Substantial evidence indicates that vehicle movement is closely linked to the spread of diseases among animal production sites. To mitigate these transmission events, vehicles undergo thorough cleaning and disinfection (C&D) procedures. However, C&D effectiveness remains an open-ended question, while the frequency of C&D between farm visits is unknown. Consequently, relying solely on vehicle C&D is insufficient to control the spread of diseases, and supplementary strategies are necessary to prevent disease transmission events via contaminated vehicles. The objective of this study was to reduce the risk of between-farm transmission through vehicle contacts by rerouting vehicles while considering C&D events and effectiveness.
Methodology: We collected movements of 654 vehicles in a pig-dense area in the U.S. and identified vehicles visiting farms, C&D, slaughterhouses, feed mills, and parking locations. Farm data was collected from enhanced on-farm Secure Pork Supply (SPS) biosecurity plans available in the Rapid Access Biosecurity application (RABappTM). We ranked and reorganized the vehicles delivering animals and feed to farms according to several conditions, including disease status of visited farms, vehicle contact network communities, C&D events, and shipment time efficiency. Using these conditions, we simulated vehicle movements for one week, indicating each vehicle was C&D after each shipment. We reconstructed the between-farm contact network by vehicle movements from observed and simulated data and compared i) the number of contacts from PRRSV-positive and PEDV-positive farms to disease-free farms and ii) contacts between farms from different network communities (group of farms densely interconnected). In addition, we calculated the frequency of vehicles visiting C&D stations and traveled distances.
Results: Implementing our rerouting system led to a substantial decrease in the median number of risky contacts among farms (Figure 1). For vehicles transporting feed, risky contacts were reduced by 42% when C&D effectiveness was 0% and 89% when C&D was 50%. For vehicles transporting pigs to market, risky contacts were reduced by 25%, with C&D effectiveness at 0% and 45% with C&D at 50%. Vehicles transporting pigs between farms only showed a remarkable reduction after C&D effectiveness above 50%, with 33% fewer risky contacts. Finally, with C&D effectiveness at 100%, risky contacts dropped below 5% for vehicles transporting feed and pigs to market, and below 37% for vehicles transporting pigs to farms. Our system also reduced the interactions between farms from distinct network communities, 17% when C&D effectiveness was 0%, and 99.9% with C&D effectiveness at 100%. Furthermore, the rerouting system increased by up to 81% in C&D visits and up to 54% in distance traveled per vehicle.
Discussion and conclusion: We showed that by rerouting vehicle movements, risky contacts among farms were reduced. Despite the potential benefit of preventing disease spread, our rerouting system increased the C&D events and the distance traveled per vehicle. Given the severe economic impact of PRRSV, PEDV, ASFV, and other endemic infectious diseases on swine production, the costs and logistics of a vehicle rerouting system require a close examination to justify the potential economic benefits of reducing disease transmission compared to continuing traditional vehicle movement schedules and C&D protocols. Ultimately, our rerouting system could be integrated into regular vehicle shipment schedule operations as an additional tool for preventing and controlling the spread of livestock diseases among farms via the indirect contact network of vehicle movements
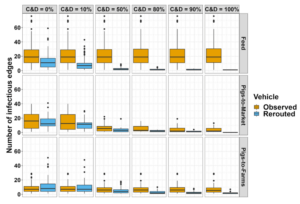
Figure 1. Infectious edges within the vehicle contact network. Box plots displaying the number of connections, named infectious edges or risky contact, from PRRSV-positive and PEDV-positive farms to disease-free farms within the vehicle contact network for one week.
Project #: 23-036 | Principal Investigator: Rodger Main, DVM, PhD | Institution: Iowa State University | Posted: 1/10/25 | Keywords: PRRSV, PEDV, Biosecurity, Temperature, Time.
Since its introduction in 2013, PEDV remains an important disease to the US swine industry, necessitating robust biosecurity measures. This study aimed to evaluate various cleaning protocols—Positive Control (no cleaning), Dry Clean – Scrape and Bake (TADD), Volume Hose Wash and Disinfect, Power Wash and Disinfect, and Negative Control—to determine their efficacy in mitigating PEDV spread from a contaminated trailer to the farm site area after simulating foot traffic between these two areas.
Livestock trailers were contaminated with PEDV-positive feces, except for the Negative Control using PEDV-negative feces. Different cleaning methods were applied, followed by a simulation to mimic real-world conditions. PEDV presence was quantified using qPCR to measure viral load on trailer surfaces and farm loading areas. Bioassay was conducted to inoculated naïve pigs with samples recovered from the farm site area after applying the treatments on the trailers. Statistical analyses, including ANOVA and Fisher’s Exact test, compared PEDV levels and positive PCR results across treatments.
Washing treatments, particularly volume hose and power wash & disinfect, demonstrated significant efficacy in reducing viral load on both trailer and farm-site surfaces. On farm-site surfaces, these methods achieved over 99% reduction in viral genomic copies compared to the positive control. This marked reduction is crucial in preventing infection of susceptible pigs and highlights the importance of effective washing protocols. The Power Wash and Disinfect method emerged as highly effective, significantly reducing PEDV levels on trailer surfaces with most samples showing negative PCR results (3 of 5). The Volume Hose Wash and Disinfect method also demonstrated substantial efficacy in inactivating the virus on the bioassay, though some residual fecal material was observed. Even though the Scrape and Bake method reduced the viral load in more than (>98%), it was not effective in terms of virus inactivation, where the pigs from four out of five replicates became infected for PEDV on bioassay. All negative control replicates were negative in the qPCR result of the surfaces sampled and remained negative on the bioassay after inoculation.
The findings from this study suggest routinely cleaning and disinfecting all market haul trailers leaving terminal points of concentration by either of the water-based trailer cleaning treatments evaluated present as an opportunity to create a step-change in the magnitude of inter-premises disease transmission associated with market haul transport and elevate the state of preparedness across the US pork industry.
Contact: [email protected]
Project #: 23-070 | Principal Investigator: Gustavo De Sousa E Silva | Institution: Iowa State University | Posted: 1/10/25 | Keywords: PRRSV, perinatal, mortalities, tongue, virus isolation, swine
Objectives: This study aimed to: 1) Compare the detection of the Porcine reproductive and respiratory syndrome virus (PRRSV) and Influenza A virus (IAV) using reverse transcription-quantitative polymerase chain reaction (RT-qPCR) in weekly oral fluids (OF) and TF from wean-to-market pigs, 2) Assess the likelihood of successful PRRSV ORF-5 sequencing from these two sample types (OF vs. TF), 3) Assess the effect of pooling TF on the RT-qPCR detection of PRRSV.
Research Methodology: This study monitored three groups of pigs from two production systems, testing for PRRSV and influenza A virus (IAV) from weaning until market. Two groups (A and B) were sourced from PRRSV-positive stable sow farms, while Group C was from a farm weaning positive pig. Groups B and C received a PRRSV-modified live virus (MLV) vaccine at weaning. Weekly, oral fluid (OF) samples from six pens and TF samples were tested for PRRSV and IAV using RT-qPCR. PRRSV-positive TF and OF samples were selected and Sanger sequenced to compare the performance of both sample types. PRRSV-positive TF samples were also serially diluted to assess the impact of pooling on PRRSV detection. The study evaluated associations between weekly pathogen detection and mortality using mixed-effects regression models, as well as diagnostic accuracy and agreement between OF and TF results. Additionally, statistical models were used to assess the cycle threshold (Ct) changes with varying dilution factors to determine the minimum dilution level at which PRRSV detection was maintained in 95% of cases.
Research Findings: There were 60 weeks across all three groups with sample submissions, OF samples were obtained in all 60 weeks, and TF samples were collected in 43 of those weeks. IAV was detected in 34.9% of OF and 30.2% of TF samples, while PRRSV was found in 67.4% of OF and 53.5% of TF samples. TF samples showed a significantly lower mean cycle threshold (Ct) for PRRSV (28.7) compared to OF samples (33.2). There was fair agreement between the RT-qPCR outcomes of TF and OF (Cohen’s kappa values of 0.26 for PRRSV and 0.24 for IAV). Out of the 22-week-matched TF and OF pairs sent for PRRSV sequencing, 45.5% of OF and 63.6% of TF samples were successfully sequenced. The higher success of sequencing in TF samples was attributed to their relatively lower Cts. For both sample types, mortality rates were significantly higher when PRRSV was detected, especially when detected alongside IAV. Additionally, dilution studies demonstrated that pooling samples could effectively increase detection probabilities, with 79% detection at a 1/7 dilution compared to 14.29% detection probability if only one day’s TF out of one week is tested for the same scenario of 1 positive day.
Industry Implications: Tongue fluid (TF) samples offer a comparable and cost-effective alternative to oral fluid (OF) samples for the RT-qPCR detection of PRRS virus and Influenza A Virus (IAV) in growing pig herds, especially when dead animals are available. Despite PRRSV and IAV RT-qPCR detection being numerically more frequent in OF, TF samples had a higher success rate for PRRSV sequencing due to their significantly lower Ct values. This makes TF a useful surveillance sample type for PRRSV in wean-to-finish barns. To maximize the diagnostic potential of this sampling approach, practitioners are encouraged to aggregate tongue tissues from all daily mortalities. As there could be weeks without mortality, postmortem TF sampling and antemortem sampling (e.g., OF) can be jointly part of a surveillance program. Furthermore, testing TF pools in weekly pools can be useful in wean-to-finish barns where budget constraints may limit testing options.
Project #: 23-051 | Principal Investigators: James F. Lowe | Institution: Lowe Consulting Ltd. | Posted: 12/16/24 | Keywords: Swine biosecurity, Trailer decontamination, Disease transmission, Epidemiological modeling, Cost-benefit analysis
Objectives: The primary goal of this study was to determine the most effective and cost-efficient way to clean market haul trailers that transport pigs between wean-to-finish farms and slaughter facilities. We aimed to understand how different levels of trailer washing impact the spread of Porcine Epidemic Diarrhea virus (PEDv) and to find the best practices that balance disease control and economic feasibility.
Research Conducted: We used computer simulations to model swine production systems under various conditions. The first scenario focused on a single production system with 24,000 sows across 8 sites. The second scenario looked at a region with 24,000 sows divided into 4 systems, each geographically related but otherwise unconnected. We ran simulations to see how washing different percentages of trailers would affect the spread of PEDv. Additionally, we tested scenarios with different PEDv prevalence levels to see how these conditions would change the effectiveness of trailer washing.
Findings: 1. Single Production System:
– Washing 100% of trailers significantly reduced the number of infected farms, with an average of 23.13 infected premises.
– Washing 60% of trailers was identified as a cost-effective strategy, achieving a significant reduction in disease spread at a lower cost of about $32,956 per farm.
2. Geographically Related Systems:
– Washing 100% of trailers in this setup reduced the average number of infected premises to 10.56.
– Surprisingly, washing 0% of trailers was found to be the most cost-effective in these systems, costing about $25,664 per farm, suggesting that extensive decontamination might not always be necessary in such segregated systems.
3. Impact of Disease Prevalence:
– In scenarios with high PEDv prevalence (20%), it was necessary to wash 80% of trailers to achieve a significant reduction in infection, at a cost of $48,957 per farm.
– In low prevalence scenarios (5%), washing trailers was not required to minimize costs, which dropped to $26,862 per farm.
What These Findings Mean for the Industry:
The study shows that cleaning market haul trailers is crucial for controlling the spread of PEDv, but the extent of washing needed can vary. For interconnected systems or during high prevalence periods, thorough washing of trailers is essential. However, in more isolated systems or when disease prevalence is low, producers might save costs without compromising biosecurity by washing fewer trailers. These insights can help producers develop tailored cleaning protocols that enhance swine health and productivity while managing costs effectively.
By adopting these findings, producers can make informed decisions about trailer decontamination practices, ensuring their operations are both economically viable and biosecure. This research provides a practical roadmap for reducing PEDv transmission and safeguarding the health of swine herds.
Project #: N/A | Principal Investigators: Montse Torremorell, Juan Mena, Ana Marcos, Marie Culhane | Institution: University of Minnesota | Posted: 10/17/2024 | Keywords: HPAI, H5N1, influenza, literature review
The recent emergence of high pathogenic avian influenza (HPAI) H5N1 clade 2.3.4.4b in dairy and the ongoing outbreaks of HPAI in commercial poultry seriously threaten the U.S. swine industry. Due to the role that pigs play in the overall ecology of influenza infections, the potential risk of H5 infections in people, and the epidemiological links between swine, dairy, and poultry, it is of the utmost importance to fully understand the risks to pigs and from pigs to other species, including humans. Of equal importance is the development of science-based strategies needed to prevent the introduction of H5N1 into pigs and contain it should incursions occur.
Project #: 23-067 SHIC | Principal Investigator: Onyekachukwu Henry Osemeke | Institution: Iowa State University | Posted: 9/27/2024 | Keywords: PRRSV, perinatal, mortalities, tongue, virus isolation, swine
Objectives: This study aimed to assess different sample collection protocols and cell lines for successfully isolating live porcine reproductive and respiratory syndrome virus (PRRSV) from perinatal piglet mortalities (stillborn piglets and piglets that died within 24 hours of birth) using tongue tissue fluids (TF).
Research Methodology: Samples were obtained from 597 perinatal mortalities in a 5,000-head PRRSV-positive breeding herd over a 4-day period. Tongue tissues were grouped into 20 batches (approximately 30 mortalities or tongues per batch). Each tongue was divided into four quarters, with each quarter randomly assigned to one of four collection protocols: 1) TF extraction from fresh tissues using Phosphate Buffered Saline (PBS), 2) TF extraction from fresh tissues using Virus Transportation Medium (VTM), 3) TF extraction in PBS after one freeze-thaw cycle (Freeze-thaw), and 4) using tongue tissue homogenate (Homogenate). This resulted in 80 samples total (20 batches x 4 protocols), all sent to a NAHLN-approved veterinary diagnostic laboratory for RT-qPCR testing. The RT-qPCR cycle threshold (Ct) values were averaged across the four protocols in each of the 20 batches, and the 10 batches with the lowest mean Ct values were selected for virus isolation (VI). Two cell lines (ZMAC and MARC-145) and one batch of primary alveolar macrophages (PAM) were tested for their effect on successful PRRSV isolation.
Research Findings: All samples tested positive for PRRSV by RT-qPCR; the average Ct values for the PBS, VTM, Freeze-thaw, and Homogenate groups were 21.9, 21.8, 22.6, and 24.8, respectively.
PRRSV was isolated successfully from tongue tissues in all groups with varying success rates. The virus isolation success rate was 22.6% in the PBS group, 12.1% in the VTM group, and 2.8% in both the Freeze-thaw and Homogenate groups. The probability of successful viral isolation was 3.1% in MARC-145 cells, 21.0% in ZMAC cells, and 4.8% in PAM cells. Mortality batches with only stillborn piglets had a 35.5% probability of successful PRRSV isolation, while batches with stillborn and dead piglets had a 1.0%.
Industry Implications: Live PRRSV can be isolated from postmortem tongue fluids. Extracting TF from fresh stillborn piglets using PBS or VTM increases the chances of successful virus isolation. The ZMAC cell line outperformed the MARC-145 cell line and PAM cells in this study. Ensuring a cold chain from sample collection until arrival at the laboratory maintains the diagnostic quality of the samples. Isolating PRRSV from aggregate samples such as TF provides several surveillance and vaccine development benefits. Virus isolation using aggregate samples allows for the efficient co-detection of multiple PRRSV strains within a herd if present, this has great advantages for surveillance and developing autogenous vaccines.
Project #: 23-078 SHIC | Principal Investigator: Mariana Kikuti, Cesar A. Corzo | Institution: University of Minnesota | Posted: 9/27/2024 | Keywords: postmortem sampling; PRRSV detection; specimen sensitivity; disease monitoring; diagnostic accuracy
Specimens collected from dead pigs are a welfare-friendly and cost-effective active surveillance. This study aimed to evaluate the accuracy of different postmortem specimens from dead piglets for disease detection, using PRRSV as an example. Three farrow-to-wean farms undergoing PRRSV elimination were conveniently selected. Samples were collected at approximately 8- and 20-weeks post-outbreak. Postmortem specimens included nasal (NS), oral (OS), and rectal (RS) swabs, tongue-tip fluids (TTF), superficial inguinal lymph nodes (SIL), and intracardiac blood. These were tested individually for PRRSV by RT-PCR. Sensitivity, specificity, negative and positive predictive values, and agreement of postmortem specimens were calculated using intracardiac sera as the gold standard. OS and SIL had the best overall performance, with sensitivities of 94.6–100%, specificities of 83.9–85.1%, and negative predictive values of 97.3–100%. TTF had high sensitivity (92.2%) but low specificity (53.9%) and positive predictive value (48.3%). While challenges in meeting sampling
targets due to variable pre-weaning mortality were noted, PRRS was detected in all postmortem specimens. OS and NS showed promising results for disease monitoring, though TTF, despite their sensitivity, had lower specificity, making them less suitable for individual infection assessment but useful for assessing environmental contamination.
Project #: 23-044 | Principal Investigator: Dr. Derald Holtkamp | Institution: Iowa State University | Posted: 8/20/2024 | Keywords: Standardized Outbreak Investigation Program, emerging disease, transboundary disease, Rapid Response Rrogram, epidemiology
Industry Summary: To protect the United States swine industry from substantial economic losses caused by emerging, or transboundary disease outbreaks, producers and veterinarians need to rapidly identify, control, and slow the spread of the pathogen. In response to events following the introduction of the porcine epidemic diarrhea virus (PEDV) into the United States in 2013, the Swine Health Information Center (SHIC) funded Iowa State University to develop the Rapid Response to Emerging Disease Program (RRP) in August 2016. The program now includes a nationwide network of veterinarians, state animal health officials or representatives, epidemiologists, and, when appropriate, federal animal health officials called the Rapid Response Team (RRT). RRT members are trained, prepared, and committed to moving within 24 hours of contact to conduct epidemiological investigations when a new transboundary or emerging disease threat occurs. The approach, methodology, form and reports were developed to systematically, consistently, and comprehensively conduct outbreak investigations for the RRP.
In 2021, SHIC funded the development of the Standardized Outbreak Investigation Program for the RRP (21-080 SHIC, 22-076 SHIC, 23). The form developed for the Standardized Outbreak Investigation Program to conduct outbreak investigations, formatted as a Word® (Microsoft, Redmond Washington) document, is available for download on the SHIC website at https://www.swinehealth.org/investigation-instrument/ (accessed May 29, 2024). A web-based application was developed for the Standardized Outbreak Investigation Program. The application was launched in January of 2024 and is available to RRT members and other veterinarians and producers to conduct outbreak investigations on sow farms.
The objective of this project was to provide ongoing management and coordination of the RRP to maintain the readiness of the RRT.
The transitional Project Coordinator, who was appointed in 2021, provided support to RRT members, assisted in conducting outbreak investigations of endemic diseases, and was available in the event of an animal health emergency where the RRT was called upon.
The principal investigator (Dr. Holtkamp), Transitional Project Coordinator (Chris Mowrer) and Dr. Kate Dion collaborated with SES Inc., who was contracted by SHIC, to develop nine training videos. to develop training for the RRT and other users of the Standardized Outbreak Investigation Program web application. The final version of these videos is expected by June of 2024.
Contact Derald Holtkamp at [email protected]
Scientific Abstract: Same as Industry Summary.
Introduction: To protect the United States swine industry from substantial economic losses caused by emerging, or transboundary disease outbreaks, producers and veterinarians need to rapidly identify, control, and slow the spread of the pathogen. In response to events following the introduction of the porcine epidemic diarrhea virus (PEDV) into the United States in 2013, the Swine Health Information Center (SHIC) funded Iowa State University to develop the Rapid Response to Emerging Disease Program (RRP) in August 2016. The program now includes a nationwide network of veterinarians, state animal health officials or representatives, epidemiologists, and, when appropriate, federal animal health officials called the Rapid Response Team (RRT). RRT members are trained, prepared, and committed to moving within 24 hours of contact to conduct epidemiological investigations when a new transboundary or emerging disease threat occurs. The approach, methodology, form and reports were developed to systematically, consistently, and comprehensively conduct outbreak investigations for the RRP.
In 2021, SHIC funded the development of the Standardized Outbreak Investigation Program for the RRP (21-080 SHIC, 22-076 SHIC, 23). The form developed for the Standardized Outbreak Investigation Program to conduct outbreak investigations, formatted as a Word® (Microsoft, Redmond Washington) document, is available for download on the SHIC website at https://www.swinehealth.org/investigation-instrument/ (accessed May 29, 2024). A web-based application was developed for the Standardized Outbreak Investigation Program. The application was launched in January of 2024 and is available to RRT members and other veterinarians and producers to conduct outbreak investigations on sow farms.
Objectives: The objective of this project was to provide ongoing management and coordination of the RRP to maintain the readiness of the RRT.
Materials & Methods: Support a Transitional Project Coordinator and investigations conducted by RRT members. Funding for this project provided support for a transitional Project Coordinator who was previously appointed in 2021. When requested by producers, RRT members were invited to conduct investigations of outbreaks of endemic diseases. The transitional Project Coordinator gathered the information needed to prepare the outbreak investigation form and coordinated the investigations with all of the relevant parties (RRT member, herd veterinarian, farm manager, etc.). The investigations provided opportunities for RRT members to practice investigations of outbreaks of endemic diseases.
Training of the RRT on the web-based industry-standard outbreak investigation instrument. SHIC contracted with SES Inc. to develop training for the RRT and other users of the Standardized Outbreak Investigation Program web application. The principal investigator (Dr. Holtkamp), Trasitional Project Coordinator (Chris Mowrer) and Dr. Kate Dion collaborated with SES to develop nine videos.
Results: Transitional Project Coordinator and investigations conducted by RRT members. The transitional Project Coordinator provided support to RRT members and assisted in conducting outbreak investigations of endemic diseases and was available in the event of an animal health emergency where the RRT we be called upon to conduct outbreak investigations. If such an event were to occur, it is expected that a full-time project coordinator would be required, as the number of investigations would increase significantly to assure the investigations were conducted quickly. Requests for RRT members to conduct SHIC funded outbreak investigations have declined. Only one request was received in 2023/2024, down from 13 in 2022/2023.
Training of the RRT on the web-based industry-standard outbreak investigation instrument. The investigators collaborated with SES, who was contracted by SHIC, to develop nine training videos. The final version of these videos is expected by June of 2024.
Discussion: The RRP aims to build the infrastructure to respond to emerging or transboundary disease threats rapidly to slow the spread of the pathogen. The value of that infrastructure depends on the RRT members’ ability to collect epidemiological information to identify hazards and characterize the geographic and temporal patterns of how the virus is spreading from one herd to another. Maintaining the readiness of the RRT is critical to realizing the value if an emerging or transboundary disease event occurs in the US.
Project #: SHIC 23-019 | Principal Investigators: Erin Kettelkamp, DVM | Institution: Swine Vet Center | Posted: 6/14/2024 | Keywords: aerosol, biocontainment, biosecurity, fan covering, PRRS, swine
Objective: The primary objective of this study was to evaluate the effectiveness of various rapidly deployable, exhaust fan cover materials in reducing airborne particles that can carry swine respiratory pathogens. These pathogens include viruses such as PRRSv (Porcine Reproductive and Respiratory Syndrome virus), PEDv (Porcine Epidemic Diarrhea virus), and IAV (Influenza A virus). By identifying effective and rapidly deployable fan coverings, we aim to enhance biosecurity measures and reduce the spread of these diseases within and between swine farms.
Research Conducted: The study was conducted at a commercial swine facility, focusing on three types of fan covers: PolyKlean™ synthetic air filter media (Blue Poly), a nylon tear-resistant fan sock (Fan Sock), and a polyethylene privacy screen material (Black Screen). These were compared to fans with no coverings (control). Airborne particle counts were measured using a handheld particle sizer to determine the reduction in particles ranging from 0.3 to 5.0 micrometers (µm). Measurements were taken at various distances from the fans to assess the effectiveness of each covering material. Weather conditions and ventilation settings were carefully monitored and recorded to ensure consistency across all sampling points.
Research Findings: The results showed that the Fan Sock was the most effective in reducing airborne particle quantities (0.7 to 5.0 µm) at 1 meter from the fan compared to the Blue Poly and control treatments, with the Black Screen treatment being intermediate. However, as the distance from the fan increased, differences in particle quantities were not observed across the treatments, resulting in no overall differences.
Implications for the Industry: These findings suggest that implementing exhaust fan coverings can be most beneficial at reducing larger air particles up to short distances (up to 1 meter) from fans. Based on the relationship between air particle size and the spread of airborne swine pathogens, additional research is warranted to understand the role of fan coverings on biocontainment. The fan sock provided better airflow and is already commonly used in the swine industry, making it a more practical option for rapid deployment during disease outbreaks to potentially improve regional biocontainment. Further research is recommended to validate these findings and explore additional biosecurity measures.
Project #: 24-015 | Principal Investigators: Pablo Piñeyro | Institution: Iowa State University | Posted: 7/9/2024 | Keywords: PCV4, In situ hybridization, Lymphoid tissue, Coinfection, PCV2, PCV3
Since PCV4 was first described in 2019, the virus has been identified in several countries in Southeast Asia and Europe. Most studies have been limited to detecting PCV4 by PCR. Thus, PCV4 has an unclear association with clinical disease. This study utilized 512 porcine clinical lung, feces, spleen, serum, lymphoid tissue, and fetus samples submitted to the ISU‑VDL from June–September 2023. PCV4 was detected in 8.6% of samples with an average Ct value of 33. While detection rates among sample types were variable, lymphoid tissue had the highest detection rate (18.7%). Two ORF2 sequences were obtained from lymphoid tissue samples and had 96.36–98.98% nucleotide identity with reference sequences. Direct detection of PCV4 by RNAscope revealed viral replication in B lymphocytes and macrophages in lymph node germinal centers and histiocytic and T lymphocyte infiltration in the lamina propria of the small intestine. PCV4 detection was most commonly observed in nursery to finishing aged pigs displaying respiratory and enteric disease. Coinfection with PCV2, PCV3, and other endemic pathogens was frequently observed, highlighting the complex interplay between different PCVs and their potential roles in disease pathogenesis. This study provides insights into the frequency of detection, tissue distribution, and genetic characteristics of PCV4 in the US.
Project #: 24-001 | Principal Investigator: Dustin Boler | Institution: Carthage Innovative Swine Solutions | Posted: 7/9/2024 | Keywords: ATP bioluminescence, disease monitoring, sow farm biosecurity, surveillance
This research evaluated the performance of three adenosine triphosphate (ATP) instruments to assess the cleanliness status of farrowing crates and the level of contamination after washing. Visual inspection to determine if a farrowing room is clean usually occurs after the invested cost of disinfectant and immediately prior to sows entering the room. At the same time, studies have demonstrated that visual inspection of farrowing rooms may be insufficient to ensure cleanliness and reduce disease transmission risk. The goals of this project were to determine the areas of the farrowing crate with the greatest surface contamination, the correlation between microbial counts and relative light units, and the number of locations that are needed to accurately determine surface cleanliness. The results from this study indicated that the areas of highest concern were the entryway floor and the sow feeder as detected both by ATP bioluminescence and coliform plate counts (CPC). Bioluminescence is a monitoring tool that should be used in conjunction with periodic microbial validation to monitor procedures for cleaning and disinfection.
Project #: 22-002 | Principal Investigators: Giovani Trevisan & Daniel Linhares | Institution: Iowa State University | Posted: 7/9/2024 | Keywords: data analysis, surveillance, preparedness, animal health threats, diagnostic
This study evaluated the use of endemic enteric coronaviruses polymerase chain reaction (PCR)-negative testing results as an alternative approach to detect the emergence of animal health threats with similar clinical diseases presentation. This retrospective study, conducted in the United States, used PCR-negative testing results from porcine samples tested
at six veterinary diagnostic laboratories. As a proof of concept, the database was first searched for transmissible gastroenteritis virus (TGEV) negative submissions between January 1st, 2010, through April 29th, 2013, when the first porcine epidemic diarrhea virus (PEDV) case was diagnosed. Secondly, TGEV- and PEDV-negative submissions were used to detect the porcine delta coronavirus (PDCoV) emergence in 2014. Lastly, encountered best detection algorithms were implemented to prospectively monitor the 2023 enteric coronavirus-negative submissions. Time series (weekly TGEV-negative counts) and Seasonal Autoregressive-Integrated Moving-Average (SARIMA) were used to control for outliers, trends, and seasonality. The SARIMA’s fitted and residuals were then subjected to anomaly detection algorithms (EARS, EWMA, CUSUM, Farrington) to identify alarms, defined as weeks of higher TGEV-negativity than what was predicted by models preceding the PEDV emergence. The best-performing detection algorithms had the lowest false alarms (number of alarms detected during the baseline) and highest time to detect (number of weeks between the first alarm and PEDV emergence). The best performing detection algorithms were CUSUM, EWMA, and Farrington flexible using SARIMA fitted values, having a lower false alarm rate and identified alarms 4 to 17 weeks before PEDV and PDCoV emergences. No alarms were identified in the 2023 enteric negative testing results. The negative-based monitoring system functioned in the case of PEDV propagating epidemic and in the presence of a concurrent propagating epidemic with the PDCoV emergence. It demonstrated its applicability as an additional tool for diagnostic data monitoring of emergent pathogens having similar clinical disease as the monitored endemic pathogens.
Project #: 23-031 | Principal Investigators: Daniel C. L. Linhares; Gustavo de Sousa e Silva | Institution: Iowa State University College of Veterinary Medicine | Posted: 7/1/2024 | Keywords: Manure; Manure pumping; PRRS; PEDV; Wean-to-finish; Bioexclusion; Biocontainment
Due to its nutritional and fertilizing value to the soil, the pig manure is spread in fields surrounding pig sites for the following grain crop season. However, manure agitation and spreading poses risks to animal health due to the gases and pathogens that may recirculate in the site and surrounding sites. Therefore, the goal of this project was to estimate the impact of manure pumping practices on PRRSV and PEDV health outcomes in wean-to-finish (W2F) pig populations. A retrospective and prospective studies were conducted separately.
The retrospective epidemiological study estimated the odds of PRRSV or PEDV outbreak occurring within four weeks after manure pumping out from the site event (exposure 1) or being located in a field receiving manure at 1-, 3-, and 5-miles from a site (exposure 2). The study population was W2F lots from one production system that pumped manure between July 2020 and December 2022. PRRSV or PEDV outbreaks (cases) were defined based on veterinarian assessment, pathogen detection in tissues, and increased mortality rate after the pumping event or receiving manure from other site. For the analyses, controls were selected to match spatially (within 6.2 miles of cases) and temporally (placement dates within a 4-week interval from outbreak dates) cases. The analyses revealed that the odds of PRRSV outbreaks events within a 4-week following pumping out of the site and spreading manure activities were higher. Additionally, nurseries had higher odds of reporting a PRRSV outbreak following pumping out activities compared to grow-finish. No associations between PEDV outbreak and manure practices were detected in this retrospective study.
The prospective study assessed the frequency of PRRSV RNA and PEDV RNA detection in pit samples from midwestern W2F barns and the likelihood of increasing PCR-positivity of pig oral fluids after manure pumping. The population of interest was wean-to-finish lots from a swine producer that pumped manure between April 2023 and December 2023. PRRSV and PEDV were tested by PCR on oral fluids and pit manure. All growing pig barns were selected based on the absence of PRRSV or PEDV before the pumping process.
The analyses revealed an increase in the odds of testing PRRSV-positive in oral fluids after pumping out manure. The PEDV positivity in manure was significantly higher than that of PRRSV, however there was no increase in oral fluids PEDV-positivity after pumping manure. Anyways, there was evidence of both PRRSV and PEDV virus in manure samples, confirming the ecological importance of manure in viral spread within and between sites.
Both studies (retrospective and prospective) showed that manure pumping practices were associated with PRRSV outbreak and spread. The odds of a PRRSV outbreak within a 4-week window were greater when the site was pumped and was in close proximity to a field receiving manure. The odds of a previously PRRSV-negative barn becoming PRRSV-positive increased significantly after manure pumping. This information enables veterinarians and producers to justify strategies for biosecurity and biocontainment associated with pumping manure out of sites or when a site is near a field receiving manure.
Project #: 23-037 | Principal Investigators: Daniel Linhares & Edison Magalhães | Institution: Iowa State University | Posted: 6/11/2024 | Keywords: market pigs; trailer sanitation; automated; data integration; compliance verification
The pilot project aimed to address the challenge of documenting truck washes between visits to slaughterhouses and return to swine barns, a critical aspect of market haul sanitation in the swine industry. The primary goal was to assess three different methods for automatically recording truck wash events and market pig deliveries at packing plants, thus, enabling producers to verify trailer cleanliness compliance. Furthermore, automated reports were produced to inform the decision-makers on the status of the trailers, and identify the non-compliance events (i.e., trailers not washed between loads to the packing plant).
The project was conducted in collaboration with one swine producer in the US Midwestern region, focusing on evaluating the feasibility and effectiveness of different technologies in recording truck-related events. Three approaches were tested: GPS tracking of trucks and trailers; a software application (APP) for automatically creating electronic tickets of washing events; and manual data collection at truck wash and packing plant sites. The research team collected and analyzed data over a specified period to evaluate the accuracy and reliability of each method.
The findings revealed that while all three methods had their strengths and limitations, GPS-based tracking showed higher accuracy in documenting truck wash events and deliveries at packing plants compared to the other methods. However, GPS-based methods were susceptible to errors such as false or duplicate events and the geofence limits are not adjusted, highlighting the importance of optimizing technology parameters to minimize discrepancies. On the other hand, despite the CleanTrailer APP having a slightly inferior performance for recording truck wash events, it provided an electronic ticket with pictures of before and after the wash, providing additional information beyond the electronic wash ticket. Despite some missed washes, the agreement between GPS data and the CleanTrailer APP was generally high, indicating the potential of automated systems in ensuring compliance with sanitation standards.
The results of this pilot study have significant implications for the swine industry, as providing producers with automated reports to monitor truck wash compliance. The scalability of the methods tested suggests broader applicability across production systems, offering a standardized approach to monitoring market haul sanitation practices. Ultimately, these findings empower producers to make informed decisions regarding truck sanitation, thereby safeguarding animal health and improving overall industry practices.
Project #: 23-071 | Principal Investigator: Dr. Michael Chetta | Institution: Talent Metrics Consulting | Posted: 5/16/2024 | Keywords: safety, biosecurity, biocontainment, wean-to-market, caretaker, mitigation, prevention, preparedness, compliance
The Swine Health Information Center has identified that caretaker motivation related to compliance with biosecurity behaviors is a priority needing to be better understood. An exploratory study was conducted to establish a baseline for worker motivation and identify possible issues within the industry that could be impacting compliance with biosecurity. This research and measurement related to motivation is the first of its kind in the industry and sets the groundwork for better understanding the primary factors influencing worker motivation and compliance.
Initial findings suggest the swine industry’s problem with biosecurity compliance is not a motivationally driven issue, and not wholly influenced in the way initially conceptualized and measured. There is strong support that biosecurity compliance is influenced by job resources (specifically supervisor support), availability of performance feedback and rewards. Additionally, the analyses suggest workers are heavily impacted in doing their work and adhering to biosecurity protocols by physical workload and demanding contact with animals.
There is reason to believe that motivation can be assessed differently and that the impact of training and measuring the implementation/effectiveness of biosecurity procedures could yield valuable insights. Continuing this research across the industry will help one of the largest industries in the US to better understand the interactions and motivations behind worker attitudes and perceptions towards biosecurity adherence and to enhance positive outcomes for employees, farms, and consumers.
Project #: 23-046 | Principal Investigator: Francisco Cabezon | Institution: Pipestone Research | Posted: 5/16/2024 | Keywords: Power-washing, robotics, cleanliness, water usage, labor
A 2,400 head wean-to-finish barn with two rooms of 1,200 head capacity (196 feet x 50 feet) with 44 pens each was used in the study. A group of nursery pigs were placed in the barn and raised until harvest. The barn was then cleaned, with one room washed using traditional manual power washer methods from a contract service, and the other room cleaned using a railed robotic power washer prototype, followed up with a manual power wash to remove any additional manure (touch-up). The trial consisted of two washing events (August 2023 and February 2024) to compare the efficacy and efficiency of an automated power washer to a manned power-washing crew, based on cleaning time, manpower time, water usage, and cleanliness rate.
In the room washed with the rail robotic power washer prototype, four rails were installed (2 on each side of the room divided by the central hallway) to cover the pen floor and side walls at a maximum height of 10 inches from the slat level. The rail robotic power washer prototype consisted of a trailer head carrying a rotary nozzle connected to a gas power washer. The trailer head was battery powered, and the speed of the trailer on the rail and the speed of rotation of the nozzle could be adjusted. Two different rotary nozzles were tested. The robot power washer with a single rotary nozzle was set to move through the rails at an average speed of 11.0 inches/min, with a nozzle rotation time cycle of 22 seconds (August 2023 data). In the case of the double rotary nozzle, the robotic power washer was set to move at an average speed of 14.8 inches/min, with a nozzle rotation time cycle of 30 seconds (February 2024 data). In both cases, the speed of the trailer head and rotation of the nozzle were adjusted to achieve 2 hits per slat.
Multiple methods were used to evaluate cleanliness (pre-wash, post-wash, and post touch-up): visual assessment, adenosine triphosphate (ATP) measurements to assess organic material, bacterial culture with dip slides, and a reverse-transcriptase real-time PCR (RT-qPCR) for rotavirus detection. There were 12 pens assessed in each room, which were equally spaced throughout the room. Five sites in each pen were assessed: the fencing, floor, wall, waterer, and feeder.
In August 2023 (single rotary nozzle test), total water usage in the robotic power washing room was 8,396 gallons in comparison to 6,211 gallons in the manual power washing room. Total washing time in the robotic power washer room was 22.1 h (13.0 h of robotic washing and 9.1 h of manual touch up washing) in comparison to 10.5 h of manual power washing in the control room. The manual washing labor time in the robotically washed room was reduced 13% (1.4 h), but total washing time was longer by 11.6 h.
In February 2024 (double rotary nozzle data), total water usage in the robotic power washing room was 10,897 gallons in comparison to 7,526 gallons in the manual power washing room. Total washing time in the robotic power washer room was 19.3 h (10.1 h of robotic washing and 9.2 h of manual touch up washing) in comparison to 13.3 h of manual power washing in the control room. In this case, manual washing labor time in the robotically washed room was reduced by 31% (4.1h) with the robot, but overall washing time was longer by 6 h.
Cleaning scores differences before and after washing were significant for each power washer method, at all sites in a pen, and in each testing. The cleanliness trend was from very dirty to clean or very clean. For the robotic power washed room, the post-wash touch-up by the manual power washing team was necessary for the median value to reach the “Very Clean” score.
More bacterial count, rotavirus presence, and ATP levels were found after the washing process for both wash methods. Power washing does not clean the barn, it is solely a means to remove debris and must be followed by a disinfection process. Power washing should be completed to the necessary level to ensure that disinfection can be performed well.
Cleaning expectations of this barn were extremely high, this could explain to some degree the long touch-up process. Robotic power washer cannot easily access the feeders. The washing crew spent considerable time washing the feeders. The number of feeders in the barn will be a limiting factor to the efficiency of the robotic power washer. The barn used for this research has a low pigs/feeder ratio (27 pigs/feeder, doubled 1-hole wet dry feeder). Another limiting factor for the automated power washer is the number of rails and their positioning. In the current study 4 rails were installed in the room. This allowed walls to be washed at a maximum height of 10 inches from the slat level, however, the wash did not cover the central hallway. Additional rails could increase the covered area by the rail power washer, but it would represent additional costs and time of operation.
Although power washing needs at facilities are time and resource intensive, this robotic power-washer prototype does not provide adequate savings in manpower or water usage, so further refinements are needed.
Project #: 23-079 | Principal Investigator: Dr. Mariana Kikuti | Institution: MSHMP/UMN | Posted: 4/17/2024 | Keywords: Porcine deltacoronavirus; epidemiology; swine health; disease occurrence; incidence
PDCoV, an enveloped RNA virus, causes atrophic enteritis in neonatal piglets, leading to diarrhea, malabsorption, dehydration, and death. The study aims to fill the gap in the current epidemiological information about PDCoV in the U.S. pig population after its emergence in 2014. Data from the Morrison Swine Health Monitoring Project (MSHMP) between January 2015 and December 2023 were analyzed, representing approximately 60% of the U.S. breeding herd. Participating herds report weekly PDCoV health status. In total, 244 PDCoV outbreaks occurred in 186 sites from 22 production systems across 16 states. Case counts peaked during winter, and incidence ranged from 0.44% in 2017 to 4.28% in 2023. For sites that experienced more than one PDCoV outbreak during the study period, the interval between outbreaks was a median of 2.11 years. The South and Midwest regions reported the majority of cases. In 2017, a shift in the spatial distribution of cases from the Midwest to the South was observed. The findings underscore the importance of continued monitoring and strengthened control measures to mitigate the impact of PDCoV in U.S. breeding herds.
Project #: 22-059 | Principal Investigator: Dr. Gustavo Machado | Institution: North Carolina State University | Posted: 3/28/2024 | Keywords:Truck Transport, Disease modeling, Contact trace, Indirect contact, Truck cleaning, Disinfection
Project #: 22-076 | Principal Investigator: Dr. Derald J. Holtkamp | Institution: Iowa State University| Posted: 3/27/2024 | Keywords: Biosecurity, hazard analysis, outbreak investigation, investigation form, swine
Sustained progress on improving the intended outcomes of biosecurity has been elusive. Improvement in outcomes for endemic diseases, such as the reduced frequency of outbreaks in sow farms, has not been observed. The rapid spread of PEDV after it was introduced into the US in 2013 and the spread of African swine fever virus (ASFV) worldwide (WOAH, 2023) are reminders of the ongoing threat of emerging and transboundary diseases. A more comprehensive, strategic and deliberate approach to identifying, assessing and prioritizing biosecurity hazards to efficiently allocate biosecurity resources is urgently needed to make sustainable improvements in the intended outcomes of biosecurity. Outbreak investigations are excellent opportunities to improve biosecurity if the aim of the outbreak investigation is to collect enough information to confidently identify, analyze and prioritize the biosecurity hazards that need to be addressed first.
In 2021, the Swine Health Information Center (SHIC) funded the development of a Standardized Outbreak Investigation Program. A working group of fourteen swine veterinarians was formed to develop terminology, the approach and an investigation form and reports for conducting outbreak investigations and reporting the findings. The Standardized Outbreak Investigation Program borrowed heavily from methods developed for the hazard analysis and critical control point (HACCP) methodology and may be used to conduct investigations on sow farms as an integrated biosecurity hazard analysis and epidemiological investigation.
A critical concept for identifying biosecurity hazards is the three failures. This concept is foundational for the standardized outbreak investigation conducted as a biosecurity hazard analysis. The three failures are 1) failure to prevent the pathogen-carrying agent from being contaminated or infected with an infectious pathogen, 2) failure to mitigate the contamination or infection of the pathogen-carrying agent, and 3) failure to prevent pig(s) in the herd from being infected with the pathogen from the pathogen-carrying agent after the pathogen-carrying agent arrives at the site. All three failures are necessary for transmission to occur. Using the three failures concept, a biosecurity hazard can be defined as any aspect of the production process that increases the probability of one or more failures. A biosecurity hazard analysis is conducted by investigating the production procedures and the pathogen-carrying agents associated with entry events to identify the biosecurity hazards.
The form developed for the Standardized Outbreak Investigation Program was originally formatted as a Word® (Microsoft, Redmond Washington) document. The limitations of the form formatted as a Word document were that 1) it was relatively difficult to use, 2) there was not a convenient way to store and access company-specific historical information collected during investigations, and 3) it was not feasible to capture the data from all users to build a secure industry-wide database that could be used to learn from the collective experience of veterinarians and producers. The aim of this project was to develop a web-based application and database for the Standardized Outbreak Investigation Program to overcome those limitations.
The Standardized Outbreak Investigation Program web application may be accessed by producers or veterinarians at https://fieldepi.org/soip/. Requests to set up a new company may be made by email to [email protected]. Companies and sites can be created in the application and investigations for several swine pathogens may be conducted for individual sites. The application includes an easy to navigate form to conduct the investigations. The form is organized into two main sections. The first section is the pre-investigation survey. It is intended to capture information before the investigation is conducted on-farm. The second section is the investigation form which is to be used to capture information during the investigation conducted on-farm and from any follow-up conducted.
Upon completion of an investigation, a detailed report and an executive summary may be viewed and saved as PDF files. The detailed investigation report includes all the information collected from the pre-investigation survey and the investigation form. The executive summary is a shorter report that summarizes the investigation’s major findings, including a table of hazard ratings for each entry event and the frequency of each entry event during the investigation period. A summary of the entry events that were rated high, including the justification for rating the event as high, is included in the executive summary. The executive summary also includes a summary of the weather during the investigation period.
Outbreak investigations are magnificent opportunities to identify and prioritize biosecurity hazards. Outbreaks are a crisis and an opportunity to learn, but learning is not guaranteed. If outbreak investigations are conducted systematically, deliberately and comprehensively to identify, assess and prioritize biosecurity hazards, the information learned will provide a return on the investment in the investigation and sustained progress can be made on the intended outcomes of biosecurity.
Project #: 22-072 | Principal Investigator: Dr. Natalia Cernicchiaro | Institution: Kansas State University| Posted: 3/22/2024 | Keywords: Japanese encephalitis, JE, JEV, risk assessment, swine
Japanese encephalitis virus (JEV), which is associated with encephalitis in humans, and reproductive and neurological illness in pigs, has expanded to areas free from disease, most recently in Australia with the invasion and expansion of JEV in eastern and southeastern states. The current study’s overarching goal is to increase awareness and build capacity to cope with the risk of a potential JEV incursion. The introduction of this virus into the United States (US) could not only impact public health, but also animal health and the food supply. To efficiently and cost-effectively manage risk, a better understanding of how and where this disease will be introduced, transmitted and spread is required. The objective of this study was to update and reassess the 2018 qualitative risk assessment implemented to estimate the risk of emergence of JEV into continental US and add information regarding transmission post-emergence, establishment and spread, by incorporating the latest scientific information, newer assumptions, data sources, recent outbreak data from Australia, and regional elements contributing to the risk, which may have been previously overlooked.
We used an established semi-quantitative risk assessment framework to evaluate the risk of introduction of JEV into seven US regions from the current region of distribution (i.e., southeast Asia and the western Pacific rim), and its subsequent spread and consequences. The regions at risk were created by grouping the 48 states of the contiguous US into five regions based on similar climate, density of domestic swine production, and presence of feral swine; due to their geographic isolation, Alaska and Hawaii were included as their own individual regions. Information utilized to update model parameters included data from an updated systematic review of the literature on vector and host competence for JEV. Our updated review assessing literature published from 2016-2022 reported a slightly higher number of articles that focused on host competence and susceptibility to JEV compared to vector competence. This shift in focus could be attributed to, in part, the realization of certain host species’ (e.g., pigs, migratory and domestic birds) high susceptibility to JEV. One striking feature explored in an experimental study, which has yet to be replicated, is the ability of JEV to be transmitted between pigs without the aid of mosquito vectors. Consequently, there is a growing recognition of the importance of controlling not only the vector, but also the virus at the animal level, particularly through vaccination and other control strategies.
Additional sources of information for our risk assessment models included global databases, scientific literature, and expert opinion obtained from our advisory group and project collaborators. Our advisory group consisted of swine producers, veterinarians, entomologists, researchers and stakeholders from the US and Australia with knowledge on commercial swine production, Japanese encephalitis virus, and the latest Australian outbreak.
Summary output parameters calculated by this risk assessment include the rate of introduction, estimated epidemic size, and the overall risk estimate. The highest risk scores for the rate of introduction were deemed moderate and associated with the entry of adult mosquitoes in cargo ships into the US south (RAR 3; AL, AK, DE, Fl, GA, KY, LA, MD, MS, NC, OK, SC, TN, TX, VA) and west (RAR 5; CA, ID, OR, WA) regions, and via eggs/larvae in imported tires into the northeast (RAR 1; ME, MA, NH, NY, VT) and west (RAR 5) regions. The largest estimated epidemic size was greater than one million host animals in the US south (RAR 3), whereas the lowest estimated epidemic size was 10 host animals in the northeast (RAR 1). In the midwest region (RAR 2; CT, IL, IA, KS, MI, MN, MO, NE, NJ, ND, OH, PA, RI, SD, WV, WI) the estimated epidemic size ranged from 10 to between 1,000 and 10,000 host animals, and in the west (RAR 5) region from 100 to between 100,000 and one million. The overall risk, which reflects the rate of introduction and economic impact of a JEV incursion into the region at risk, was very high for the south (RAR 3), moderate for the west (RAR 5), and very low for the northeast (RAR 1) and the midwest (RAR 2) regions.
In summary, based on the results from this risk assessment, the US south region (AL, AK, DE, Fl, GA, KY, LA, MD, MS, NC, OK, SC, TN, TX, VA) should be prioritized for preparedness to mitigate the impact of disease, whereas the west (CA, ID, OR, WA) and northeast (ME, MA, NH, NY, VT) should also be prioritized for surveillance activities towards early disease detection.
As with any risk model, the results of this study are entirely dependent on the data used and the assumptions made. In this assessment, we supplemented the limited data on JEV transmission (e.g., the role of vector-free transmission among pigs and of non-ardeid birds and wildlife in the JEV transmission cycle in US), spread, and persistence, with information gathered from the latest Australian JEV outbreak of 2021-23, existing and new available scientific evidence, and expert opinion. This qualitative risk assessment provides a framework necessary for decision-makers to assess the risk of JEV introduction and potential impacts in the US, which is crucial to prepare for the threat of an emerging disease and protect the swine industry from suffering huge economic losses.
Project #: 22-094 | Principal Investigator: Dr. Cesar A Corzo | Institution: University of Minnesota | Posted: 3/20/2024 | Keywords: Morrison Swine Health Monitoring Project, MSHMP, disease monitoring, PRRS, PEDV, PDCoV, Senecavirus
Objective 1: Monitor trends in pathogens incidence and prevalence – PRRSv, PEDv, PDCoV, Senecavirus and central nervous system associated viruses continued to be monitored and each maintained the historical pattern. We also investigated the relationship between the PRRS RT-PCR positivity rate between breeding and growing pigs finding a correlation; however, because no spatial information and prrs virus information was accounted for we need to interpret this information with caution, thus we are unable to conclude that the increase in PRRS positive rate in breeding herds is due to an increase in positivity rate in growing pigs. In addition, we also assessed the frequency of PRRS outbreaks in breeding herds managing mortality either through composting, incineration, or rendering. Numerical differences were detected but not enough evidence was found to conclude that one method is better than the other one and more analyses are needed. Regarding PEDV, we characterized the time positive herds required to stabilize the herd and concluded that those infected during the epidemic phase of the disease in the US took longer (e.g., 9 weeks) to reach stability than those infected during the endemic phase.
Objective 2: To conduct prospective monitoring of PRRSv sequence evolution and impact – We characterized the epidemiological situation of an emerging virus (L1C-124) that was thought to be a virulent and fast spreading one. Even though the virus did disseminate, it did not reach a speed and magnitude of other recent viruses. The clinical impact was also characterized but when compared to the recent L1C-144 we can conclude that it was not significantly different.
Objective 3: To expand participation of producers to allow for all to be involved – During 2022-2023 we have added 4 production systems. Our database now has 1,274 sow farms housing approximately 3.8M sows, 3,318 growing pig farms with 12.2M pig spaces and 48 boar studs totaling 12,799 boars. Our first publicly available MSHMP website has been completed, officially launched in Q3 of 2023, and has been continuously updated. This website was designed to optimize MSHMP output dissemination within both the participant and industry communities. The website represents a significant milestone in our mission to enhance collaboration and communication within the swine industry. The website has been designed with ten comprehensive sections, including Home, About, History, People, Reports, Outreach, Ongoing Projects, News, Resources, and Contact Us. The website has been accessed by individuals from 41 countries with the highest number of users from the US, China and Spain.
Project #: 23-045 | Principal Investigator: Dr. Rahul K. Nelli | Institution: Iowa State University | Posted: 3/20/2024 | Keywords: Japanese encephalitis virus, JEV, RT rt-PCR, JEV detection
Japanese Encephalitis Virus (JEV) is a severe disease affecting pigs and humans. It is spread mainly by mosquitoes and is endemic in many parts of Asia and the western Pacific regions. However, its recent outbreak in Australia posed a renewed threat to the US pig industry and public health. Therefore, it is crucial to have a reliable way to detect JEV in pigs before it causes an outbreak. The current project aims to detect JEV rapidly using viral RNA and Reverse Transcription real-time polymerase chain reaction (RT-rtPCR) assays. This project established RT-rtPCR assays for JEV and its five genotypes (G-1, -2, -3, -4, -5) and compared the newly established test with a previously published assay. The findings from this project suggest that the newly developed RT-rtPCR assay is more specific and can detect all five genotypes accurately and efficiently. It also had a low detection limit, meaning it could detect very small amounts of the virus. Validation of genotype-specific RT-rt PCR assay and evaluation of the assay performance on known status clinical samples in collaboration with Australia is ongoing. Our project developed a novel and reliable test for JEV that can help the pig industry and public health authorities monitor and prevent JEV outbreaks.
Project #: 22-065 | Principal Investigators: Marcelo Almeida, Alyona Michael | Institution: Iowa State University | Posted: 2/5/2024 | Keywords: App, surveillance, oral fluids, nasal swabs, tonsil scrapings, survivability
After an outbreak of Actinobacillus pleuropneumoniae (App) serotype 15 in central IA in 2021/22 three initiatives were put in place to investigate the atypical behavior of this event. Serological surveillance was performed in 19 of 30 sow farms supplying pigs to sites experiencing outbreaks. Most sow farms (16 of 19) were App antibody negative, suggesting that at least part of the finishing sites may have had a lateral introduction of the bacteria. Evaluation of individual (nasal swabs and tonsil scrapings), population (oral fluids) and environmental samples were collected longitudinally over 6 weeks starting 3 weeks post the reported App 15 outbreak in a finisher to evaluate post-outbreak persistence dynamics. Tonsil scraping had the highest detection rate of App by PCR, while nasal swabs only detected positive pigs in the first 3 weeks of sampling. Oral fluids could detect positive pens during the 6 weeks, but missed pens with positive pigs. Environmental samples were sporadically positive by PCR. Finally, a comparative evaluation of three different App strains (serotype 15 outbreak and historical strain and serotype 1) were compared in liquid media and solid surfaces at different temperatures for up to 7 days. Warmers temperatures (25°C and 37°C) decreased the survivability of all three strains and colder temperatures (-20°C and 4°C) were favorable to survivability. Differences were observed in liquid media (higher survivability in horse serum-NAD compared to water and fecal slurry) and solid surfaces (concrete > rubber > stainless steel). This information adds to App ecology and epidemiology knowledge and may help inform about diagnostic, monitoring and surveillance, and biosecurity decisions.
Project #: 21-133 | Principal Investigator: Chad Paulk | Institution: Kansas State University | Posted: 12/19/2023 | Keywords: Chlorine Dioxide, Feed Mill Decontamination, Flushing, Formaldehyde, Viral contamination, Paulk
This project set forth to understand the best flushing, thermal processing, and decontamination techniques for a feed mill contaminated with porcine epidemic diarrhea virus (PEDV), porcine reproductive and respiratory syndrome virus (PRRSV), and Seneca Valley virus 1 (SVV1); furthermore, infectivity of samples collected after utilizing these techniques was evaluated using a swine bioassay. Formaldehyde flushes (either liquid or dry) were most effective at reducing viral concentrations in both the feed and the environment; however, no method eliminated viral RNA from the environment. Although PEDV and SVV1 RNA were detected via PCR, upon inoculation to pigs, treatments failed to cause infection which provides evidence of successful mitigation. However, PRRSV viral particles were able to cause replication in pigs when introduced from both feed and dust samples even when the inoculum samples were negative for PRRSV via PCR. It is unknown why PRRSV RNA was not consistently detectable via PCR, but the bioassay suggests more research is needed, especially regarding PRRSV mitigation. Thermal processing reduced the detectable RNA in both feed and the environment for all viruses. Replication of SVV1 and PEDV was not observed in the swine bioassay, but PRRSV replication was observed in the post-thermal processing feed. Increased temperatures may be required to inactivate PRRSV if contamination were to occur pre-thermal processing. The complete facility decontamination (removal of organic matter with heated pressure washing, disinfection with 1% Virkon, disinfection with 5% household bleach, environmental heat held at 140°F for 48 hours) was the only decontamination treatment where PEDV, PRRSV, and SVV1 RNA was non-detectable after completion of all steps. The chlorine dioxide and heat treatments reduced the detectable quantity of RNA, but viral particles were still detectable across mill surfaces. Although PEDV and SVV1 RNA was detected via PCR, only PRRSV replication was observed in the swine bioassay from the chlorine dioxide treatments and one heat treatment. Overall, chemical flushing, thermal processing, and facility decontamination reduce the viral RNA presence, but more research is needed to understand how these techniques affect the infectivity of these viruses.
Project #: 23-018 | Principal Investigator: Dustin Boler, Bailey Harsh | Institution: Carthage Innovative Swine Solutions, University of Illinois | Posted: 11/14/2023 | Keywords: Truck wash biosecurity, ATP bioluminescence, disease monitoring, surveillance
This research evaluated the performance of two adenosine triphosphate (ATP) instruments to estimate cleanliness in livestock trailers. The results of the ATP instruments were compared to aerobic plate counts (bacterial contamination) to determine if ATP bioluminescence could be used as an indicator of trailer cleanliness without the need for visual inspection. ATP is a source of cellular energy that is present among all living organisms. This includes viruses and bacteria that remain after a commercial trailer cleaning. Normally, trailer cleanliness is determined visual evaluation by a person that inspects the trailer to determine if it is free of organic material and suitable to return to a farm. However, studies have demonstrated that visual inspection of cleaned transport trailers may be insufficient to ensure cleanliness and reduce disease transmission risk because viruses and bacteria are microscopic in nature and cannot be seen by the human eye. Further, visual inspection to determine if a trailer is clean usually occurs after the invested cost of propane to dry the truck has occurred. ATP bioluminescence uses a chemical reaction where a swab is used to detect the presence of ATP. The more ATP that is present, the greater the chemical reaction. This technology uses the same chemical process a firefly uses to illuminate. When ATP is exposed to the enzyme a light is produced. The more ATP that is present the brighter the light. The intensity (brightness of the light) is measured in relative light units (RLU). A greater RLU indicates more ATP and reduction in overall cleanliness. So, this technology can be equated to the brightness of a firefly. The brighter a firefly glows; the more ATP is present. In this case, the brighter the swab glows, the more ATP is present, the more potential microbial contamination is present. The goals of this project were to determine the areas of the trailer with the greatest surface contamination, the correlation between microbial counts and RLUs, and the number of locations that need to accurately determine surface cleanliness.
This project evaluated livestock trailers at two commercial trailer washes. One of those locations included trailers that were known to transport livestock from a farm known to be PRRS or PEDv positive. In total, 100 livestock trailers were tested usings biolumiomters to determine the amount of ATP that remained after a commercial trailer wash. Protocols are in place to prevent people from entering a livestock trailer after it has been cleaned to prevent contamination. For this technology to be adopted into practice, locations inside the trailer that are accessible from outside the trailer must be evaluated. This trial evaluated the back door flush gate (BDFG), rear drivers side access door (RSAD), the belly flush gate (BFG), belly side access door (BSAD), nose side access door (NAD).

Figure 1 Back door flush gate. An area of the trailer that is indicative of overall trailer cleanliness.
The results from this study indicated that the areas of highest concern sampled in this study were the nose access door and the back door flush gate as detected both by ATP bioluminescence and APC. A key finding of this research was that nose access door was the area least likely to be adequately cleaned, but only a few trailers actually had nose access doors. Nearly all of the trailers evaluated had a back door flush gate and therefore makes it a logical place to swab a livestock trailer to determine the overall cleanliness.
The ability of ATP to be used as an indicator of trailer cleanliness was dependent on the instrument used, with the 3M machine being more closely correlated with bacterial contamination. Swabs were also collected to determine the presence of PEDv, but all swabs were negative. These data suggest that ATP bioluminometers can be used in livestock trailers to quickly determine the general cleanliness of the trailer without the need for a visual inspection. Bacterial swabs to determine APC levels should also be used to determine the effectiveness of the cleaning protocol.
Project #: 22-071 | Principal Investigator: Dr. Monteserrat Torremorell | Institution: University of Minnesota | Posted: 9/12/2023 | Keywords: whole-genome, long-read sequence, viruses, target enrichment
Viral co-infections on swine farms are common and frequent, contributing to aggravated disease outcomes and impairing economic profit and animal well-being. In addition, due to high mutation rates, the co-circulation of viruses within swine herds facilitates the emergence and re-emergence of new variants, impairing disease investigation, control, and prevention. Despite their significant cost and health relevance, viral co-infections are understudied in part due to limitations in sequencing techniques able to generate full-length sequences of multiple viruses from field samples. Furthermore, the detection and genomic characterization of novel variants are still a challenge, and many questions remain about the dynamics and interactions of co-circulating viruses in swine herds. To advance our understanding of viral co-infections, we have developed a workflow called “TELSVirus”, or “Target-Enriched Long-read Sequencing of Virus” that enables the real-time detection and genomic characterization of multiple viral pathogens from a single sample in a relatively short turnaround time (~24 h). The main objective of this study was to apply the “TELSVirus” workflow to porcine oral fluid samples to detect and characterize genomes of target viral pathogens. Overall, this method allowed us to detect a high prevalence of co-circulating yet understudied viruses, including porcine bocavirus, porcine sapelovirus 1, and porcine astrovirus 2 and 4; while also allowing for detection of viruses with known production impacts, such as porcine reproductive and respiratory syndrome virus (PRRSV), influenza A virus (IAV) and rotavirus. These viruses were often detected in the same oral fluid sample, and the high sequencing coverage afforded by TELSVirus allowed for robust variant analysis. We also demonstrated that TELSVirus’ limit of detection for PRRSV and IAV is comparable to that of qPCR, while providing increased genomic information. Based on these results, TELSVirus has the potential to support real-time surveillance of endemic and emergent viruses, while also improving our understanding of co-circulating viruses; their genetic diversity; and ultimately how they impact swine health and production.
Project #: 21-139 | Principal Investigator: Pablo Pineyro | Co-investigator: Marcelo Almeida | Institution: Iowa State University | Posted: 8/28/2023
Porcine circovirus type 3 (PCV3) was initially described as affecting swine in 2016, and since its first description, PCV3 has been detected globally and retrospectively in cases of pigs of all ages. It has been associated with a range of clinical presentations, including reproductive failure, porcine dermatitis and nephropathy syndrome, multisystemic inflammation, respiratory disease, enteric disease, and subclinical infection. However, for many of these clinical presentations, the causative role of PCV3 remains questionable, and only a few are real clinical problems. The current diagnosis is based on viral detection by PCR and occasional in situ confirmation by in situ hybridization. However, PCV3 has been detected in clinical and subclinically infected animals. Therefore the lack of correlation between the viral detection and the presence of lesion(s) or specific clinical signs make it imperative to redefine and improve the PCV3 case definition that can be used to trigger field investigation and better control the PCV3 infection.
One of the objectives of this study was to establish a Ct PCR value that correlates with the presence of lesions compatible with PCV3 infection. Our retrospective study showed that there is a positive correlation between Ct values and the presence of lesions. Cases confirmed by histopathology with lesions consistent with multisystemic inflammation showed Ct values below 30. In addition, the presence of PCV3 in these cases was confirmed by in situ hybridization, adding diagnostic value to the combination of histopathology and PCR detection.
In this study, the evaluation of samples that could be used for surveillance or subclinical detection included oral fluids, processing fluids, and serum. Most of these samples were not linked to specific clinical problems. Evaluation of the Ct value distribution demonstrates that a high proportion of the sample presents Ct values above 32. The implication of these findings needs further evaluation. These sample matrices could indicate viral shedding, vertical transmission on sow units, or viremia in grower-finisher. However, the clinical implication of positive results on these sample matrices cannot be extrapolated to assess the presence of PCV3-associated disease.
The evaluation of submissions of reproductive failure associated with PCV3 showed that Ct values on cases confirmed only by PCR or a combination of PCR and histological evaluation do not differ. These results suggest that PCR on fetal tissue could be sufficient to confirm causation. However, a high proportion of cases showed coinfection with PRRSV and PCV2. The Ct values on these coinfection cases showed a positive correlation suggesting that these viruses have a synergist effect with a higher PCV3 viral load when found in association with PRRSV and PCV2.
We conclude that the diagnosis of PCV3-associated disease should be based on a combination of diagnostic tools, including lesions of multisystemic inflammation, significant PCR Ct values lower than 30, and additional confirmation by direct detection methods. The sample used for surveillance and subclinical evaluation could be used as a proxy for viral circulation in sow herd and grower pigs but cannot be correlated with multisystemic inflammation or reproductive failure.
Institution: University of Minnesota | Posted: 7/2019
Vitamins are essential nutrients required by swine to optimize health, productivity and well-being. The U.S. pork industry is dependent on vitamins manufactured in China because there are limited, and in some cases, there are no other country of origin options to meet industry volume demands. Initial studies have provided emerging evidence that the African Swine Fever virus (ASFv) can survive in choline chloride, but not vitamin D3. However, it is unknown if this virus can survive in other vitamins. The risk of ASFv or other Foreign Animal Diseases (FAD) being introduced from China into the U.S. through vitamin imports appears to be low, but the impact of introduction is high.
Project #: 22-070 | Principal Investigator: Dr. Natalia Cernicchiaro | Institution: Kansas State University | Posted: 7/14/2023 | Keywords: swine, Japanese encephalitis, rapid systemic review, transmission
The United States (US) is considered a susceptible region with great potential for the introduction of the Japanese encephalitis virus (JEV) given the availability of competent mosquito vector species (e.g., Culex, Aedes spp.), susceptible maintenance avian hosts (e.g., ardeid, water birds), intensive travel and trade activities to and from Japanese encephalitis (JE)-affected countries, similar climatic and environmental conditions to epidemic countries, no active JEV surveillance in place, and large populations of susceptible swine which can serve as amplification hosts. Of note, is the considerable geographical expansion shown by the JEV in the last decades, including the recent emergence of a new genotype in the eastern and southeastern Australian states. The ongoing risk of the emergence of JEV underlines a need for increased understanding of the biology of this virus, components and dynamics of transmission, and the environmental factors necessary for incursion and establishment.
With funds provided by the Swine Health Information Center (SHIC), a team of researchers at the College of Veterinary Medicine, Kansas State University, led by Dr. Natalia Cernicchiaro, associate professor of veterinary epidemiology, and in collaboration with researchers at the United States Department of Agriculture, National Bio and Agro-Defense facility and the National Feral Swine Damage Management Program, appraised scientific literature to synthesize existing evidence on the role of domestic and feral swine in the transmission of the JEV, and to identify knowledge gaps that can pinpoint areas amenable for future research. From an initial 3,183 article abstracts obtained from electronic databases and hand searches of pertinent literature, a total of 228 articles were deemed relevant and their data extracted for inclusion in this review.
Typically, JEV infection is initiated following the bite of an infected mosquito, although recent evidence indicates that direct contact between pigs may also play a significant role in transmission. Although vector-borne remains the most efficient means of transmission, recent findings indicate that direct pig-to-pig transmission may occur primarily via oronasal fluids, yet additional research is needed to ensure replication of those findings and further evaluate its implications for pathogenesis and spread. Considering vectored transmission, JEV replicates in the skin and reaches the circulatory system through the lymphatic system. In the blood, the virus can be detected as early as 1-day post-infection (dpi) and can persist for 4 to 5 days, regardless of the portal of entry. Virus titers can reach concentrations of 105 50% tissue culture infectious dose/mL (or 106 plaque forming units/mL) between 1- and 5-dpi. Once pigs are exposed to the virus, on average it takes 3 to 6 days, and up to 21 days, to observe clinical signs. Amplification and persistence mechanisms, as well as the effect of genotype in infectiousness, transmission and pathogenesis in swine need to be better elucidated.
Disease is generally mild with a neurologic or reproductive presentation depending on the age and sex of the pigs. Although maternal antibodies can confer protection for longer than four months under field conditions, naïve piglets can manifest neurologic signs including ataxia and tremors which can progress to wasting disease or mortality. Reproductive disorders including stillbirths, abortions, mummified fetuses, and delayed farrowing can occur when sows are infected before 60 to 70 days of gestation. Orchitis, reduced sperm motility and temporary infertility have been reported in boars.
Serological testing followed by virus isolation for confirmation is recommended for diagnosis. During outbreak investigations, serial collection of serum samples in pigs is used to display an increasing titer against JEV. Different tissues are amenable for virus isolation including spleen, liver, brain, sera, cerebrospinal fluid, tonsils, and oronasal fluids. In fetuses, stillbirths or neonates, virus may be isolated or detected from brain, tonsil, spleen, placenta, thoracic or abdominal stillborn fluids. Serologic tests that demonstrate antibodies in fetuses also are useful in diagnosis.
Given no specific therapy is available for treating JEV infection in pigs, control either via mitigation of reproductive losses by vaccination or application of biosecurity practices is critical. Articles describing biosecurity practices for control and risk management at the farm level were very limited. That includes the application and effectiveness of mosquito control and other pest management practices. Vaccination against JEV is the most effective measure for controlling JEV in humans and pigs. Although there are no licensed JEV vaccines for pigs in the US, live attenuated vaccines have shown higher efficacy than inactivated vaccines in natural infection and challenge studies. Vaccinating the breeding stock can control viral amplification and reproductive disease in swine. Before the mosquito season starts, young gilts and boars can be vaccinated twice with a 14 to 21 days interval. In endemic areas, growing pigs are also often vaccinated.
Evidence on risk factors for JEV morbidity pertaining to pigs and their production environment, genetic, sex, and breed differences, as well as management practices at the farm, barn or animal levels, among others, were infrequent to not existent. Although some studies reported the use of pigs as sentinels of infection, the design, implementation and effectiveness of surveillance programs in pigs was rarely described. Even though no peer-reviewed articles were retrieved on preparedness and response efforts related to pigs, national response plans for the animal and human health sectors from governments or other official entities are available from some countries where JEV is present. Of note, are all the resources and educational materials produced in response to the most recent JEV outbreaks by the Australian Government, Department of Health and Aged Care, including the Joint National JEV Outbreak response plan published in June 2023 (JEV outbreak plan). Economic assessments associated with swine, either domestic or feral, and JEV transmission, at the regional, country, state or farm levels were not identified with our search. Similarly, articles discussing impacts or implications of JE in pigs or swine operations were not available.
Pigs have a unique role in the JEV transmission cycle. Not only are pigs implicated in sustaining transmission and spillover to other hosts, but they can also exhibit clinical signs of reproductive and neurological disease, and, although not confirmed in natural infection studies yet, can develop persistent infection of their lymphoid and nervous tissues. Knowledge of the dynamics of the disease process, viral transmission mode, portals of entry and exit, control and treatment options, as well as potential impacts of the disease in swine production, which were appraised by this team and provided in this review, may inform researchers, stakeholders, and policy makers on effort prioritization and development of preparedness and response measures.
Project #: 20-174 | Principal Investigator: Maria Sol Perez Aguirreburualde | Institution: University of Minnesota | Posted: 12/21/22 | Keywords: swine diseases, global surveillance, hazards, preparedness, awareness
Since 2018, the continuous expansion of ASF through Asia, Europe, and the Americas has kept raising concerns in the industry, resonating with the events of 2013, when a devastating epidemic of porcine epidemic diarrhea (PED) virus caused far-reaching losses. Those events demonstrated the importance of developing systems to provide situational awareness to stakeholders in near-real time to coordinate actions between government agencies and the industry with the ultimate objective of preventing or at least mitigating the impact of disease epidemics. The swine industry is vulnerable to the introduction of pathogens, and their variants, from which the US is currently free. We have developed a private- public-academic partnership to support a system for near real-time identification of hazards that will contribute to the mission of assessing risks to the industry. Identified hazards were shared monthly with swine practitioners and the government to help increase the country’s awareness and preparedness. Ultimately, the system has kept contributing to identifying and early detection or creating awareness of key stakeholders to support current prevention and mitigation strategies for the introduction of foreign pathogens into the US.
Project #: 21-109 | Principal Investigator: Montserrat Torremorell | Institution: University of Minnesota | Posted: 12/1/2022 | Keywords: biosecurity, biocontainment, swine, technologies, airborne
Among all infectious agents, airborne pathogens are the most difficult to control and in today’s agricultural settings, airborne animal diseases are the most difficult to contain. This is especially relevant for swine reared in confinement and in controlled ventilated environments where exhaust fans help transport particles from inside to outside of the facility. We conducted a review of technologies directed at removing or/and inactivating pathogens for their application to swine farms and found that only air filtration solely consisting of fibrous filters is an established and implemented technology. There are also industrially and medically implementable technologies named electrostatic precipitators and ultraviolet-C systems (both in-duct and upper room), that they are commonly implemented in other industries but are less prevalent in livestock management. Nonetheless, because they have demonstrated scalable performance in aerosol particle removal and/or bioaerosol inactivation, they merit discussion and consideration towards agricultural biosecurity. Lastly, there are emerging technologies, consisting of more recently developed technologies, typically at the laboratory or bench scale that have not been tested at the scales (building sizes and flow rates) required for agricultural biosecurity and are hence several developmental steps away from direct implementation. Lastly, there are protocols and practices ready to be used at the farm level (e.g. wind breaks, covering of fans, movement of infected animals to other sites, depopulation and reduced ventilation) that conceptually, could be employed in the face of a disease outbreak, but their direct impact on reducing the emission airborne pathogens needs to be evaluated and quantified.
Project #: 21-116 | Principal Investigator: John M. Drake | Institution: University of Georgia | Posted: 9/15/2022 | Keywords: bacterial associations, BRT machine learning, spillover risk, subject matter experts, Sus scrofa domesticus, survey
A collaboration between SHIC and the Center for the Ecology of Infectious Diseases (CEID) at the University of Georgia examined spillover risk of bacteria from wild North American mammal species into the U.S. swine herd.
The first phase of the project consisted of a data-driven horizon scan which identified 102 bacteria species hosted by 127 North American wild mammal species that have a measurable chance of associating with domestic pigs. These bacteria species were further analyzed and assigned a propensity score based on their predicted ability of spilling over from their wild mammal hosts into domestic swine. A classification of either novel or known was also assigned based on whether or not these species had a known association with pigs documented in academic literature.
The second phase of the project involved a qualitative approach through the use of a survey of subject matter experts (SMEs). The goal of the survey was to rank high-risk bacteria species to assess their potential impact on the swine industry. Five industry professionals completed the survey and identified 16 bacteria species as being of high impact to the swine industry, four of which are not yet known to infect domestic swine: Anaplasma bovis, Clostridium botulinum, Klebsiella pneumoniae, and Yersinia pestis. Additionally, 7 bacteria were ranked as having a high-risk of antimicrobial resistance, and 5 bacteria were ranked as having a high human outbreak potential.
Future steps involve continued efforts to investigate pathways for potential spillover events, such as shared habitats between wildlife and domestic swine, or pathogen overlap. Additionally, different bacterial strains, serovars, and variants of bacteria species of concern must be closely examined to increase predictive power. Bacteria account for a high degree of morbidity and mortality in swine, causing huge losses to the swine industry and zoonotic risks. Domestic pigs often acquire these pathogens (either directly or indirectly) from wild animals, creating the need for assessments such as these to prioritize bacteria species for targeted prevention strategies. Industry professionals across disciplines hold valuable insights crucial to informing risks related to emerging diseases. Their collective knowledge based on years of expertise fills a gap in the absence of a comprehensive database on the pathogenicity of swine-associated bacteria. A lack of published academic literature also fuels the need to engage SMEs to develop a systematic process for identifying bacterial species of concern to the swine industry, and this research represents a summary of such an endeavor.
The benefit of this collaboration to the industry is that bacteriologists, diagnosticians, and other SMEs will have enhanced information needed to prevent, prepare, and respond to emerging diseases and their potential impact on swine health, welfare, and market.
Project #: 21-113 | Principal Investigator: Gustavo Silva, DVM, MS, PhD | Institution: Iowa State University | Posted: 9/15/2022 | Keywords: PRRSV, PEDV, biosecurity, temperature, time
Decontamination of fomites coming into sow farms using foggers at supply entry rooms is commonly used to mitigate associated risks. Although this practice has been established, recent research has begun to question the effect of this method to inactivate pathogens, especially in complex situations where pathogens may be shielded by organic material or blind spots (Kettelkamp et al., 2019; Leuck et al., 2020).
Objective: The primary objective of this study is to evaluate the efficacy of temperature and time for inactivating porcine reproductive and respiratory syndrome virus (PRRSV) and porcine epidemic diarrhea virus (PEDV) on experimentally contaminated surfaces commonly found at supply entry rooms in swine farms.
Materials and methods: The PRRSV MN184, PRRSV 144 L1C variant or PEDV Viruses were used. The surfaces were diamond plate aluminum and cardboard tested at four temperatures (68 ºF, 86 ºF, 104 ºF and 122 ºF) in combination with six holding times (15 minutes, 60 minutes, 6 hours, 12 hours, 24 hours, and 36 hours). Once the surface temperature reached the desired temperature, the coupons were held for the designated holding time. The negative controls remained at room temperature for 36 hours and the positive controls remained at room temperature for 15 minutes. Three replicates for each treatment were performed and each coupon was inoculated with 2 mL of virus or 2 mL of media for negative controls. Virus titration was performed in each sample separately after the holding time. Regression models and Weibull curves were built to assess the impact of temperature and time on virus inactivation.
Discussion: Under the conditions of this study, PRRSV 144 L1C variant was inactivated from aluminum surfaces by heating coupons to 86oF for 12 hours and for cardboard surfaces by heating the coupons to 86oF for 6 hours. Regarding PRRSV MN184, virus inactivation was possible at 86oF after 24 hours on aluminum and at 104oF after 12 hours on cardboard. PEDV inactivation was achieved at 86oF after 6 hours on aluminum and at 86oF after 12 hours on cardboard. Virus was inactivated after 15 minutes and 1 hour at 122oF in aluminum surfaces for PEDV and PRRSV 144 L1C variant, but not for cardboard.
Overall, regardless of the virus tested on this study, to reduce the risk of virus introduction at swine farms through a supply entry room, it is recommended that materials should achieve 860F, and remain at that temperature for 24 hours. Other options, would be to increase the temperate to 104oF or 122oF but decreasing the holding time for 12 hours.
Project #: 20-078 | Principal Investigator: Hiep Vu | Institution: University of Nebraska-Lincoln | Posted: 7/1/2022 | Keywords: ASFV, ASF, pen-side test, lateral flow, PCR, early detection
African swine fever (ASF) is a devastating viral disease of domestic pigs with mortality rate that can approach 100%. ASF control mainly relies on strict biosecurity that involves movement restriction, quarantine, and compulsive depopulation of affected herds. Rapid and reliable detection of ASFV infected pigs is critical for the control of ASFV. One desirable property of a diagnostic tests is the capacity to detect viral infection, especially during the incubation time, when the infected animals have not displayed any clinical signs. In this study, we evaluated performance of three pen-side tests for ASFV detection: one PCR test for detection of viral genomic DNA and two lateral flow tests for detection of viral antigens.
The first objective was to determine the time from infection to the earliest detection. Ten pigs were experimentally infected with an ASFV strain. Whole blood and oral swab samples were alternatively collected from five pigs every other day post-infection (dpi) and tested with the three pen-side tests. The pen-side PCR test detected infected pigs starting from 2 dpi when using whole blood and 3 dpi when using oral swabs. It consistently detected infection until the end of the study (10dpi). The antigen test detected infected pigs starting from 3 dpi and no longer detected infection at 10 dpi when using whole blood. The antigen test did not work well when tested with oral swabs. Compared with the reference laboratory real-time PCR test, the pen-side PCR test exhibited 97.8% sensitivity and 100% specificity. The antigen had 100 specificity but only 47.8% sensitivity, mainly because it failed to detect infection from samples collected early or late after infection.
The second objective was to evaluate the diagnostic performance of the tests with samples collected from the field. Whole blood and oral swabs were collected from 205 pigs: 34 positives and 171 negatives as determined by the reference laboratory real-time PCR. All pen-side tests had 100% specificity, regardless of the sample types tested. The sensitivity of the pen-side PCR test was 88.2% and 70.4%, respectively when tested with whole blood and oral swab. The sensitivity of the antigen test 50% and 11.11% respectively when tested with whole blood and oral swabs.
In summary, the results of this study show that the PCR pen-side test has better performance than the antigen test as it can detect infected pigs earlier and for a longer duration after infection than the antigen test. Additionally, the pen-side PCR test works with both whole blood and oral swabs while the antigen test work with only whole blood.
Project #: 21-114 | Principal Investigator: Ganwu Li | Institution: Iowa State University | Posted: 5/25/2022 | Keywords: porcine mobillivirus, virus isolation, real-time RT-PCR, virus isolation, epidemiology
Paramyxoviruses that are known to naturally infect swine include porcine rubulavirus, Menangle virus, Nipah virus, and porcine parainfluenza virus. There are reports of less well-characterized paramyxoviruses associated with central nervous and respiratory disease in pigs. However, none of these viruses are classified in the genus Morbillivirus. In the spring of 2020, the Veterinary Diagnostic Laboratory at Iowa State University received twenty-two porcine fetuses (neonatal mortality, stillbirths, mummified fetuses, and fetuses with moderate autolysis) from six litters submitted from Mexico for diagnostic investigation. PCV2, PCV3, PRRSV, PPV1, and Leptospira sp. were not detected by qPCR or RT-qPCR in any litter. Metagenomics sequencing identified a new virus in the genus of Morbillivirus: Porcine Morbillivirus (PoMV). Other currently known members in the genus Morbillivirus, including measles virus (MeV), rinderpest virus (RPV), peste des petits ruminants virus (PPRV), canine distemper virus (CDV), phocine distemper virus (PDV), cetacean morbillivirus (CMV), and feline morbillivirus (FeMV), are highly contagious pathogens and can cause serious human and animal diseases. Therefore, there is an urgent need to determine if this new virus associated with swine fetal death is present and, if yes, its prevalence in the U.S. swine population. A total of 450 clinical samples (brains, lungs, and spleens from neonatal mortalities, stillbirths, and mummified fetuses) were collected from all over the United States and subjected to PoMV rRT-PCR assay. Our current assessment has not found PoMV in the U.S. swine populations tested thus far. In addition, virus isolation would be valuable for future characterization of PoMV (diagnostics, prevention, pathogenicity, and pathogenesis investigations). Isolation of PoMV in various cell lines (MARC-145 (a clone of the MA-104 cell line, ATCC CRL-2378), MDCK (ATCC NBL-2), PK-15 (ATCC CCL-33), ST (CRL-1746), Vero (ATCC CCL-81), BHK-21 (CCL-10), LLC-MK2 (CCL-7), ZMAC (ATCC PTA-8764), and primary porcine kidney cells using available PoMV PCR-positive tissue homogenates was attempted in this study. Unfortunately, a PoMV isolate that could efficiently replicate in cell culture has not yet been obtained.
Project #: 20-073 | Principal Investigator: Derald Holtkamp, DVM, MS | Institution: Iowa State University College of Veterinary Medicine | Posted: 5/21/2022 | Keywords: PRRSV, PEDV, PDCoV, wean-to-market, biosecurity
The swine industry has committed considerable effort to researching and improving biosecurity practices in swine breeding herds; however, attention to biosecurity in the wean-to-market phase of production lags. Events or characteristics of premises that are more frequently associated with introducing PRRSV and porcine enteric coronaviruses in wean-to-market premises are not well understood compared to the swine breeding herds.
The objective of this study was to detect the introduction of wild-type PEDV, PDCoV, TGEV, and PRRSV into groups of growing pigs and to associate the timing of the introductions with the frequency and timing of pathogen-carrying agent entry events.
Seventy-five groups of growing pigs were enrolled in the study originating from sow farms that were consistently weaning pigs that were negative for PRRSV (AASV Category II-vx or IV) and coronaviruses, including porcine epidemic diarrhea virus (PEDV), porcine deltacoronavirus (PDCoV) and transmissible gastroenteritis virus (TGEV). Eight pen-level oral fluid (OF) samples were collected from each group of pigs at placement and every two weeks until the end of the wean-to-market phase for all the groups enrolled in the study and tested via PCR for PRRSV, PEDV, PDCoV, and TGEV. Pathogen-carrying agent entry events and biosecurity characteristics for each group were tracked on a two-week basis for each group of pigs paired with the diagnostic results. The categories included premises demographics, people movement, vehicles/deliveries, carcass disposal, air and water entry, swine movements, manure removal, entry of other animals, and disease status.
Overall, 15.5% of groups became positive for PEDV, 26.4 % became positive for PDCoV, and 97.3% became positive for PRRSV. No cases of TGEV were detected.
During the nursery phase, groups that tested PEDV positive had a higher number of average wean pig delivery events, feed deliveries, propane and fuel deliveries, new supply entries, caretaker visits, and maintenance performed outside the barns than nursery premises that tested negative for PEDV. PDCoV positive nursery groups had a higher average number of wean pig delivery events, feed deliveries, propane and fuel deliveries, new supply entries, transferred supply entries, caretaker visits, maintenance performed inside the barns, maintenance performed outside the barns, visits by management, veterinarians, and other visitors compared to nursery groups that tested negative. Entry of new supplies and maintenance performed inside the barns were more frequent for nursery groups that tested positive for PRRSV than negative groups.
PEDV positive finisher groups had a higher average number of feeder pig delivery events on clean trailers, market pig removals, feed deliveries, propane and fuel deliveries, caretaker visits, maintenance performed outside the barns, and visits by management, veterinarians, and other visitors compared to PEDV negative finisher groups. PDCoV positive finisher groups had a higher average number of feeder pig deliveries, market pig removals, feed deliveries, propane and fuel deliveries, new supply entry, and caretaker visits compared to PDCoV negative finisher groups. PRRSV positive finisher groups had a higher average number of feeder pig deliveries, market pig removals, cull pig removals, feed deliveries, rendering removal, propane and fuel deliveries, transferred supply entry, caretaker visits, and visits by management, veterinarians, and other visitors compared to PRRSV negative finisher groups.
Project #: 21-074 | Principal Investigator: Ganwu Li | Institution: Iowa State University | Posted: 4/30/2022 | Keywords: Streptococcus equi subspecies zooepidemicus (S. zooepidemicus), whole genome sequencing analysis, genomic island, virulence gene, pahtogenicity, epidemiology
High mortality events due to Streptococcus equi subspecies zooepidemicus (Streptococcus zooepidemicus) in swine in the United States were firstly reported in Ohio and Tennessee in September and October 2019. One and a half year later (February, 2021), two-year-old adult sows from a production system in Indiana experienced increased sow death loss with 66 deaths in a 2400 sow on production herd within a six-week period. To investigate if the Indiana outbreak isolates were similar /different to those isolates from Ohio and Tennessee outbreaks, whole genome sequencing analysis was performed. Four outbreak isolates from Indiana were genetically distant to those isolates caused high mortality events in Ohio and Tennessee in the spring of 2019, while closely related to an S. zooepidemicus isolate from a horse in Iowa, suggesting that more than one strain of S. zooepidemicus could cause high mortality events in the United States. The genome sequence of the Indiana outbreak isolate was further closed using Nanopore sequencing, comparative genomic analysis was performed, and genomic islands and putative virulence genes were identified. Two genomic islands (GI-3 and GI-13) were identified specifically only in the Indiana outbreak isolates, thus could serve as the biomarker for the diagnosis of this particular strain. In addition, M-like protein gene (szM) and the Fic domain-containing protein gene (bifA) were positive in those Ohio and Tennessee outbreak isolates, but absent from the Indiana outbreak isolates. Identification of biomarkers of bacterial virulence will significantly improve our response to future possible outbreaks caused by S. zooepidemicus.
View Full Report: As published in peer-reviewed journal October 2023
Project #: 20-178 | Principal Investigator: Cassandra Jones | Institution: Kansas State University | Posted: 1/7/2022 | Keywords: swine, epidemiology, feed safety, porcine delta coronavirus
Two feed mills and three breed-to-wean facilities were investigated after being diagnosed with porcine deltacoronavirus (PDCoV) with initial suspicion that feed manufacture and delivery processes were involved in disease transmission. Both feed mills were audited and environmental samples collected in areas that were deemed high risk for virus contamination. All breed-to-wean facilities had PDCoV detected as would be expected, while the only positive samples for enteric coronaviruses associated with feed mills were feed delivery trucks. These results indicate that feed delivery surfaces can help spread virus during an ongoing disease outbreak and must be considered when determining the outbreak origin.
Project #: 20-172 | Principal Investigator: Cesar A Corzo | Institution: University of Minnesota | Posted: 2/11/2022 | Keywords: monitoring, emerging, PRRS, PEDV, trends
Objective 1 of 4: Monitor trends in pathogens incidence and prevalence – PRRSv, PEDv, PDCoV, Senecavirus and central nervous system associated viruses continue to be monitored. The 2020-2021 season fortunately ended with the fourth lowest PRRSv breeding herd cumulative incidence (25.8%) during the last 11 years of monitoring. During this year we continued to monitor the emergence and dissemination of a new PRRS variant that caused production losses in the Midwest and changed the seasonality pattern historically observed with PRRS with several associated outbreaks occurring during spring 2020. PEDv continued to be present at a low incidence level as the cumulative incidence remained at 3.5%. Our exploratory data analysis showed that reporting PRRS outbreaks and manure pumping activities are associated as 40% of the breaks occurred within 30 days of this event regardless of type of manure storage.
Project #: 21-115 | Principal Investigator: Scott Dee | Institution: Pipestone Research | Posted: /30/2022 | Keywords: swine, feed, soybean meal, extended storage, viral diseases, temperature
Viruses of veterinary significance such as African swine fever virus, foot and mouth disease virus, Pseudorabies virus and Classical swine fever virus are known to survive for extended periods in plant-based feed ingredients imported into North America. To mitigate risk, high risk ingredients, such as oil seed meals, are stored in designated facilities for extended periods under controlled environmental conditions to minimize viral infectivity prior to use. The results from this study suggest that a storage period of 30-days at a temperature of 23.90 C are required to reduce virus infectivity in plant-based feed ingredients such as soybean meal. The outcomes of the study are important, since previous storage periods for feed were based only on mathematical half-life calculations, not controlled studies using live pathogens and representative conditions. We now have for the first time, scientifically sound data based on the use of infectious agents and representative conditions to advise farmers, feed mill operators, federal officials, regulatory and practicing veterinarians, and feed industry leadership on how long and at what temperature to store feed and feed ingredients, to minimize risk. Hopefully, this information will enhance the application and efficacy of Responsible Imports protocols as we collectively work to manage the global risk of feed.
Project #: 20-155 | Principal Investigator: Chad Paulk | Institution: Kansas State University | Posted: 11/2/2021 | Keywords: soy imports, transboundary shipping, foreign animal disease, organic soybeans, soybean meal
Soy-based products are known to pose a viable risk to US swine herds because of their ability to harbor and transmit virus. This project aimed to evaluate soy imports into the US as a whole and from foreign animal disease positive (FAD-positive) countries to determine which products are being imported in the highest quantities and observe potential trends in imports from FAD-positive countries. Import data were accessed through the United States International Trade Commission website (USITC DataWeb) and summarized using R (version 4.0.2, R core team, Vienna, Austria). Twenty-one different Harmonized Tariff Schedule (HTS) codes were queried to determine quantities (metric tonnes, MT) and breakdown of different soy product types being imported into the US from 2015 to 2020. A total of 78 different countries exported soy products to the US in 2019 and 2020 with top contributors being Canada (546,467 MT and 481,497 MT, respectively), India (397,858 MT and 430,621 MT, respectively), and Argentina (122,116 MT and 79,471 MT, respectively). Soy oilcake (582,273 MT) was imported in the largest quantities, followed by organic soybeans (270,194 MT) and soy oil (134,436 MT) for 2020. Of the 78 countries, 46 had cases of FAD reported through the World Organization for Animal Health (OIE) World Animal Health Information Database (WAHIS). Top exporters of soy products to the US from FAD-positive countries in 2019 and 2020 were India (397,858 MT and 430,621 MT, respectively), Argentina (122,116 MT in 2019), and Ukraine (40,293 MT and 56,392 MT, respectively). The risk of FAD introduction to the US through soy imports can fluctuate based on where FAD outbreaks are occurring, shipping methods, and end usage of products. A system to monitor these factors could help make future decisions about trade and risk of FAD introduction to US swine herds. Based on information generated from this project the following frequently asked questions (FAQ) and best practices for importation of soy products were compiled below.
Investigators: Fangfeng Yuan1,2, Xingyu Yan1,2, Brandi Feehan2, Rui Guo2, Yanhua Li2, Giselle Cino2, Ying Fang1, 2*, Douglas Marthaler2*
Institution: 1University of Illinois, 2Kansas State University | Posted: 9/15/2020 | Keywords: porcine sapelovirus, emerging swine virus, PSV monoclonal antibodies, PSV ELISA
An emerging porcine sapelovirus was isolated in a diagnostic specimen from a US swine farm, designated as PSV KS18-01. Full-length genome sequence was obtained through next-generation sequencing. Phylogenetic analysis showed that the virus is more closely related to two Japanese strains but is distantly related to two known US strains. PSV specific diagnostic tools were developed, including the monoclonal antibodies again VP1 and VP2, and a VP1-VP2 antigen-based indirect ELISA. Using this assay, the dynamic response of PSV antibody was investigated in a group of post-weaned pigs that naturally exposed with PSV. The availability of the PSV isolate (KS18-01) and the specific diagnostic reagents and assays provide important tools for PSV control and prevention.
Project #: 20-081 | Investigators: Luis G. Giménez-Lirola, Neeraja Venkateswaran | Institution:Innoceleris LLC. and Tetracore Inc. | Posted: 2/24/2023 | Keywords: Bayesian latent class analysis, ELISA, PCR, African swine fever, diagnostic test, evauation, Vietnam
In the absence of effective African swine fever virus (ASFV) vaccines, infection prevention and control through diagnostic testing and quarantine is critical. Early detection and differential diagnosis of ASFV infections increase the chances for successful control of this devastating disease. However, the interpretation of the ASF diagnostic results can be complicated due to the complex epidemiology of the disease, and its unspecific and highly variable clinical presentation, i.e., same strain producing a wide range of clinical forms. The objective of this proposal was to evaluate the performance of ASFV serum/oral fluid indirect ELISA (iELISA) (collaborative work between Innoceleris LLC. and Tetracore Inc.) for surveillance and monitoring of ASFV outbreaks in commercial farms in Vietnam. For this cross-sectional field study, our field team at Hanoi University collected a 398 paired serum/oral fluid samples, individually collected from each animal, which included 100 samples from 34 ASF-acute farms, 98 samples from 47 ASF-chronic farms, and 200 samples from 20 ASF-negative farms. The samples were tested by Tetracore ASFV iELISA and real-time PCR (qPCR). As expected, the detection rate by qPCR (74% serum; 69% oral fluid) was higher than by ELISA (16% serum; 11% oral fluid) in acute farms due to that most of the animals did not seroconvert yet. Contrary, in chronically affected farms, the detection rate of the ELISA was higher (72% serum; 57% oral fluid) than the qPCR (56% serum; 34% oral fluid). However, when we combined both qPCR and ELISA, the detection rate of ASFV positive animal increased in acute (75% serum; 74% oral fluid) and particularly in chronic farms (85% serum; 74% oral fluid). All serum samples from negative farms were negative by both ELISA and qPCR (100% diagnostic specificity) while, for oral fluids, we obtained 100% and 99% diagnostic specificity for qPCR and ELISA, respectively. The high diagnostic specificity of the tests is particularly important for ASF surveillance. Absence of false positives avoid false alarms and disruption in production, and lack of confidence in the tests/surveillance system. This unprecedented study show that there are no single best diagnostic approach for ASFV surveillance and demonstrate that the combined use of the Tetracore qPCR and indirect ELISA tests and serum/oral fluid sampling, increase efficiency of ASF disease surveillance.
Project #: 20-077 | Investigator: Marie Culhane | Institution: University of Minnesota | Posted: 11/1/2021 | Keywords: African swine fever virus, boar stud, semen, risk assessment
An ASF outbreak in the United States (US) will have a significant impact on the swine and allied industries. This proactive semi-qualitative Risk Assessment (RA) was conducted to evaluate the risk movement of liquid-cooled boar semen (semen) during an African Swine Fever (ASF) outbreak in the US swine industry will result in the spread of ASF virus (ASFv) to other premises with swine. Risks of ASFv spread associated with the movement of semen originating from a Monitored Premises within, into, and outside a Control Area were evaluated as part of a public-private partnership between the US swine industry, the College of Veterinary Medicine at the University of Minnesota (UMN), and the Swine Health Information Center. When completed, reviewed, and cleared, the RA can be used to support permits for the safe and timely movement of fresh boar semen during an ASF outbreak. This assessment is applicable to US swine production and considers the implementation of standard boar stud and semen production practices, enhanced biosecurity recommendations (EBRs) proposed by the Secure Pork Supply (SPS) plan (December 2017), and industry-agreed upon targeted mitigation measures determined by the Boar Stud Working Group (WG).
This RA is limited in scope and intended to identify ASFv transmission pathways associated with semen movement and assess the corresponding likelihoods of carrying ASFv from an infected boar stud premises and causing an infection on off-site sow or gilt breeding facilities during an ASF outbreak in the US, despite the implementation of all standard preventive measures as well as outbreak-specific targeted and/or enhanced measures. This RA will ultimately provide the framework necessary for decision-makers to both quickly assess the effectiveness of preventive measures as they pertain specifically to semen movement and also implement a permit system to allow boar studs with no known infection to move semen into, within, and outside of the ASF outbreak Control Area.
The risk evaluation is based on the likelihood of the presence of infected or contaminated semen products. The likelihoods evaluated include 1. A boar stud facility in a Control Area becoming infected with ASFv; 2. Detecting infected boars on the origin premises prior to semen movement; 3. Semen products becoming contaminated prior to movement off the boar stud; 4. Semen products becoming contaminated in transit by fomites; and 5. ASFv release during transportation of infected or contaminated semen. The likelihood that other susceptible pigs can be exposed to ASFv and become infected as a result of insemination or shipment will be evaluated in cooperation with a swine breeding WG as the proactive RA continues.
The overall findings, which require further evaluation, review, and clearance at the conclusion of the RA process, are that the risk that movement of liquid, cooled boar semen from a boar stud premises that qualifies as a Monitored Premises inside a Control Area to an off-site sow gestation or farrowing facility within or outside of a Control Area during an ASF outbreak results in the infection of susceptible swine at the destination premises is likely to be low to moderate if boar studs follow the Enhanced Biosecurity Recommendations [(EBRs) – as outlined in the Secure Pork Supply (SPS) Plan] and as agreed upon by the WG. In addition, as tactical, targeted biosecurity measures are developed with WG consultation, additional mitigations, meant to be targeted EBRs, and, assuming that these additional, strategic, targeted EBRs will be implemented as mitigation measures, the risk will likely be decreased.
This work is an evolving product-specific risk assessment that will be reviewed and updated as necessary before and during an outbreak to incorporate the latest scientific information, preventive measures, and analytical methodology. Updates and revisions to risk assessments are advisable and necessary as new data become available. The opportunity to update risk assessments to address the limitations of previous analytical methods and data might have effects on the risk ratings and outcomes. If the Incident Command System is activated in response to an ASF outbreak, APHIS (and Incident Command staff) will review this risk assessment with respect to the situation in order to assess industry requests for the movement of boar semen.
Principal Investigators: Encheng Sun, et. al. | Institution: Harbin Veterinary Research Institute | Posted: 10/28/2021 | Keywords: African swine fever virus, geotype I, virulence, pig, China
The Georgia-07-like genotype II African swine fever virus (ASFV) with high virulence has been prevalent in China since 2018. Here, we report that genotype I ASFVs have now also emerged in China. Two non-hemadsorbing genotype I ASFVs, HeN/ZZ-P1/21 and SD/DY-I/21, were isolated from pig farms in Henan and Shandong province, respectively. Phylogenetic analysis of the whole genome sequences suggested that both isolates share high similarity with NH/P68 and OURT88/3, two genotype I ASFVs isolated in Portugal in the last century. Animal challenge testing revealed that SD/DY-I/21 shows low virulence and efficient transmissibility in pigs, and causes mild onset of infection and chronic disease. SD/DY-I/21 was found to cause necrotic skin lesions and joint swelling. The emergence of genotype I ASFVs will present more problems and challenges for the control and prevention of African swine fever in China.
Project #: 20-103 | Principal Investigator: Giovani Trevisan | Institution: Iowa State University | Posted: 10/8/2021 | Keywords: data analysis, surveillance, preparedness, animal health treats, diagnostic data
In 2017 SHIC funded the Domestic Swine Disease Surveillance project, currently housed under the Swine Disease Reporting System (SDRS) initiative. The purpose of this proposal was to keep the Domestic Swine Disease Surveillance program ongoing and further develop a capability to improve sustainability over time. The specific aims included a) keep updated the aggregated database with diagnostic data from the participating laboratories, and provide monthly PDF reports to SHIC as well as keep updated the online interactive dashboards; b) enhance the sustainability of the project by migrating the report-building mechanism to an automated R Markdown technology; c) improve data exchange capability between the Data warehouse project, the Domestic Swine Disease Surveillance program, and other SDRS programs by migrating the data visualization platform to Tableau.
During the time of this project, the database was consistently updated with data from participant laboratories. The database was continuously updated and now contains data from more than 950,000 distinct submissions tested by PCR for the five U.S. porcine endemic agents, i.e., porcine reproductive and respiratory syndrome virus (PRRSV), porcine epidemic diarrhea (PEDV), porcine deltacoronavirus (PDCoV), transmissible gastroenteritis (TGEV), and Mycoplasma hyopneumoniae. Interactive online dashboards with filtering capabilities, e.g., age category, specimen, geographic region, were kept alive, updated, and are available on the project website (https://www.fieldepi.org/sdrs). Monthly PDF reports have been consistently produced and releases every first Tuesday of the month. Monthly PDF reports are published at the project webpage, SHIC website (https://www.swinehealth.org/domestic-disease-surveillance-reports/), and distributed by email to more than 270 registered receivers from 129 organizations/institutions from 7 countries (US, Canada, Mexico, Brazil, Chile, Germany, and Spain).
In July 2020 (SDRS report # 29), monthly PDF reports started to be produced using the R Markdown technology and gained a new face. An R Markdown script has written and allowed the generation of reports in a standard and consistent format. The R Markdown script was codified to automate as much as possible the process of generating and update images, formatting, and description for the monthly detection changes under each agent page. As an ongoing real-time project, there is still the need for staff to interpret the finding, debugging, communicate results with the advisory group, gather feedback, and compile the final report, including the input and feedback from the Advisory group. The usage of an R Markdown script has enhanced SDRS sustainability by reducing the time needed and errors in compiling the final monthly PDF final report.
Interconnection between databases and different visualization and analytical tools are key elements for project sustainability. The need to efficiently process and interconnect the database with the R Markdown and visualization platforms like Power BI and Tableau requested a redesign of the SDRS database structure. The SDRS has been built using a building block approach. Initially, SDRS started reporting PRRSV detection from Iowa and Minnesota VDLs and later on added South Dakota and Kansas VDLs and additional data for the enteric coronavirus and M. hyopneumoniae. Initially, each agent’s database was kept separated. A restructuring was done to compile, combine, and store all data into a single database. This redesign allowed the database interconnection with the R Markdown to generate PDF monthly reports, analytical tools like R and SAS, and business intelligence tools, like Power BI and Tableau, for data visualization.
Additionally to the proposed scope of this project, the SDRS team has consistently produced monthly audio and video reports distributed on the project website, Youtube Channel, Swine Cast platform, and LinkedIn. Also, during the last year, SDRS has been actively engaged in providing information and support to the U.S. swine industry when it went into a crisis due to the activity of contemporary and emergence of a new PRRSV strain threatening the U.S. swine health. Early detection is a key component for containing the spread of emerging or re-emerging animal health threats. More than 110 monitoring algorithms are currently implemented in the SDRS background to detect early changes in the pattern of agent detection.
This collaborative project funded by SHIC has real-time capabilities for data collection, analysis, and sharing of swine health data and information to protect and enhance the health status of the United States swine herd. SDRS is a real-time coordinated and largely representative national swine disease monitoring program that provides VDL information in efforts to minimize disease threats current and future impact. The SDRS is the only publicly available source of swine health information from U.S. VDLs. It is also the only source of information on pathogen activity in all age groups (from boar studs to grow-finish pigs). The sharing of information on endemic and endemic re-emerging diseases affecting the swine population in the U.S. has and continues to assist veterinarians and producers in making informed decisions on disease prevention, detection, and management.
Project #: 21-065 | Principal Investigator: Diego G. Diel | Institution: Cornell University | Posted: 10/5/2021 | Keywords: feed, biosecurity, nucleic acid extraction, PRRSV, SVA, PEDV
Feed biosecurity has been an area of significant interest to the swine industry. Early studies with porcine epidemic diarrhea virus (PEDV) suggested that the virus may have been transmitted through feed. Recent experimental evidence confirmed that PEDV, SVA and FMDV can indeed be transmitted through contaminated feed that is ingested naturally by susceptible pigs. One way of reducing the risk of pathogen transmission through feed is to test feed ingredients and feed before they are introduced into farms and fed to pigs. This would only be possible if sampling and nucleic acid extraction methods would allow efficient detection of pathogens in feed. In this study we focused on comparing the performance of three commercially available nucleic acid extraction kits (CORE, IndiMag, MVP II). These kits were tested in samples that were spiked with PRRSV, SVA and PEDV and that were previously collected as part of a transportation study and tested in another VDL. Our results show that the Core extraction kit outperformed the other two kits evaluated in the present study and previously used in another VDL that originally had tested the samples. Overall samples extracted with the Core kit presented lower Ct value (at least for PRRSV and SVA) and a higher sensitivity when compared to samples extracted with MVPII or the IndiMag, One of the key issues that remains to be addressed in future studies is the sampling method to be used for large volumes of feed or feed ingredients.
Project #’s: 18-137 and 18-211 | Investigators: Leonardo C. Caserta1,2, Jessica C. G. Noll1,2,Aaron Singrey1,Megan C. Niederwerder3,Scott Dee4,Eric A. Nelson1,Diego G. Diel1,2 | Institution: 1South Dakota State University, 2Cornell University, 3Kansas State University, 4Pipestone | Posted: 9/1/2021 | Keywords: feed biosecurity, half-life, ingredients, Senecavirus A, SVA, swine diseases
Animal feed and feed ingredients have recently been investigated as sources of pathogen introduction to farms and as a potential source of infection to animals postconsumption of contaminated feed. Survival of several viruses for a prolonged period has been demonstrated in feed. Here, we determined the rate of decay of Senecavirus A (SVA) in swine feed ingredients as a function of time and temperature and established half-life estimates for the virus. Select feed ingredients were spiked with a constant amount of SVA (105 median tissue culture infectious dose 50) and incubated at 4, 15 and 30◦C for up to 91 days. Virus viability and the presence of viral RNA were assessed in samples collected over time. At the three different temperatures investigated, dried distillers’ grains with solubles (DDGS) and soybean meal (SBM) provided the most stable matrices for SVA, resulting in half-lives of 25.6 and 9.8 days, respectively. At 30◦C, SVA was completely inactivated in all feed ingredients and in the control sample, which did not contain a feed matrix. Although virus infectivity was lost, viral RNA remained stable and at consistent levels throughout the experimental period. Additionally, the ability of SVA to infect swine via ingestion of contaminated feed was investigated in 3-week-old, weaned pigs. Animals were provided complete feed spiked with three concentrations of SVA (105, 106 and 107 per 200 g of feed) and allowed to naturally consume the contaminated feed. This procedure was repeated for three consecutive days. Infection of pigs through consumption of contaminated feed was confirmed by virus neutralization assay and the detection of SVA in serum, feces and in the tonsil of exposed animals by real-time reverse transcriptase PCR. Our findings demonstrate that feed matrices are able to extend the survival of SVA, protecting the virus from decay. Additionally, we demonstrated that consumption of contaminated feed can lead to productive SVA infection.
Project #: 18-194 | Investigators: Carolina Stenfeldt1,2, Miranda Bertram1, Haillie Meek1,3, Ethan Hartwig1, George Smoliga1, Megan Niederwerder2, Diego Diel4, Scott Dee5, Jonathan Arzt1 | Institution: 1Plum Island Animal Disease Center, 2Kansas State University, 3Oak Ridge Institute for Science and Education, 4Cornell University, 5Pipestone | Posted: 7/6/2021 | Keywords: contamination, feed, foot-and-mouth disease, foot-and-mouth disease virus, pig
Important work examining the role of contaminated feed as a vector for transmission of foot-and-mouth disease virus (FMDV)was published in July 2021. Specifically, the study performed by researchers from ARS/USDA at Plum Island, evaluated the potential risk of incursion of FMDV into naïve pig herds through contamination of feed. Per the study report, the goal of the project was the assessment of the infectiousness (viability) of FMDV in commercial whole pig feed and pig feed ingredients. Additionally, the researchers, led by Drs. Stenfeldt and Arzt, determined the dose required to infect pigs through natural feeding behavior. Finally, the project looked at the ability of select commercially available feed additives to reduce infectivity of contaminated feed. “While comparable research investigating the potential biosecurity risks of imported feed exists for other viral pig pathogens (Dee et al., 2018; Niederwerder et al., 2020; Niederwerder et al., 2019), this is the first comprehensive evaluation of the risk of FMDV infection of pigs through ingestion of contaminated feed under controlled experimental conditions,” wrote the authors.
Project #: 18-142 | Principal Investigator: Christa Goodell | Institutions: Boehringer Ingelheim Vetmedica GmbH, Ingelheim | Posted: 6/2021
Vietnam has lost ~6 million pigs to ASFV since the first reported outbreak in February 2019. The national swine herd has returned to 88.7% of its pre-ASFV level, but the risk of ASFV recurrence is high because of new and on-going ASFV outbreaks (Report #VM2021-0042, USDA & GAIN, May 11, 2021).
Because of the increased value of pork in Vietnam due to reduced supply, standard ASFV control measures have centered around a modified “test-and-remove” or “Tooth Extraction” protocol. A common “Tooth Extraction” protocol for a sow farm is to remove any sow exhibiting clinical signs compatible with ASF (taking a whole blood sample for ASFV PCR testing) plus the two sows in stalls on each side of the index (clinical) animal (Yaros, Leman, 2019).
The objective of this study was to test the efficacy of the “Tooth Extraction” protocol.
For each “ASFV event” (identified by farm caregiver), whole blood samples were collected from the index sow plus 14 animals in gestation stalls on each side of the index sow (see Figure 1). Samples were tested for ASFV DNA by real-time ASFV PCR within 24-hours of arrival to the laboratory. The proportion of positive animals was analyzed as a function of the gestation stall distance from the index sow.
Figure 1. Blood sampling protocol around a suspected ASF clinical sow in gestation†‡
†F0=index sow, F1= direct/closest contact neighbors, F2= indirect contact neighbors; A= ”down” row, B= “up” row
‡(The rationale for the sampling distribution was due to an assumption drinking water flowed down a common water trough)
Results. 766 whole blood samples from 52 ASFV events were collected and tested for ASFV DNA by PCR. 85 samples were positive for ASFV DNA by PCR.
Key learnings:
• In 17 (33%) of the 52 events, the index sow and 14 neighbor sows were ASFV PCR negative.
• In 35 (67%) of the 52 events, the index sow was ASFV PCR positive.
– Among allsows detected as ASFV PCR positive, 39 (78%) were located outside of the index animal and her direct contact neighbors (F0, F1A1, F1B1). Thus outside of the index animal and her contact neighbors (F0, F1A1, F1B1), 10% of all animals tested were ASFV PCR positive, compared to the direct contact animals alone (F1A1, F1B1), representing 18% of the all animals tested.
– If the index sow and 4 direct/closest contact neighbors were removed (F0, F1A1, F1B1, F2A1, F2B1), there was a 50% probability that additional ASFV PCR positive (but unidentified) sows remained.
• ASFV DNA was detected in blood from sows showing no clinical signs.
The results of this study suggest “Tooth Extraction” is not sufficient to eliminate ASF from a pig farm.
Project #: 20-175 | Principal Investigators: Dr. Ben Hause and Chun-Ming Lin | Institution: South Dakota State University | Posted: 6/17/2021 | Keywords: porcine parvovirus, porcine respiratory disease complex, tissue microarray, metagenomic sequencing, in situ hybridization , update title: Investigation of the Role of Porcine Parvovirus 2 in Porcine Respiratory and Reproductive Diseases
Discovered in 2001, porcine parvovirus 2 (PPV2) is prevalent in swine in countries worldwide. While it is commonly detected in swine samples, the clinical significance of infection is unclear. We recently identified PPV2 present at high levels in the lung of a pig with pneumonia, prompting this investigation into the role of PPV2 in respiratory disease. Here we report that PPV2 was detected in 39% of lungs submitted for routine diagnostic testing. There was a significant positive correlation between PPV2 viral load and pig age. Histologically, PPV2 was mainly detected in alveolar macrophages in 28% of lungs with interstitial pneumonia, with PPV2 viral load tending to correlate with the number of macrophages in the lungs. While co-infections with PPV2 and established swine respiratory pathogens influenza A virus and porcine reproductive and respiratory syndrome virus were commonly detected, assessment of viral loading and tissue locations showed no obvious association between PPV2 and other viral pathogens. Relatedly, in a third of PPV2 positive lungs analyzed by metagenomic sequencing, no other pathogens were identified. Together, these results suggest that PPV2 may play a primary role in porcine respiratory disease.
Project #: 19-154 | Principal Investigator: Daniel C. L. Linhares, DVM, MBA, PhD | Institution: Iowa State University | Posted: 2/9/2021 | Keywords: biosecurity, vulnerability, scoring, risk, swine, breeding herd, PRRS
PRRS continues to plague the swine industry with 20-30% of herds breaking annually. These outbreaks cost the swine industry and its producers $664 million every year. Swine producers continue to offer efforts to increase biosecurity with the intention to provide the best welfare for their pigs and produce a wholesome product for consumers. The industry needs the ability to better predict risk of a PRRS outbreak as well as to evaluate the level of biosecurity in their farms. The objective of this study was to measure and benchmark the relationship between key biosecurity aspects and PRRS outbreaks in breeding herds, while validating a short biosecurity screening survey (44 questions). The survey captured data on herd demographics, PRRS outbreak history, frequency of high-risk events, surrounding swine density, transport biosecurity practices, carcass disposal, and people movement. Three machine-learning algorithms were used to identify key biosecurity factors and practices associated with PRRS outbreaks. The most accurate, based on ability to explain variability between farms in the frequency of reported PRRS outbreaks, was selected. Moreover, positive predictive value (PPV) from the models were utilized as a biosecurity score. We investigated the correlation between PPV and the reported frequency of PRRS outbreaks.
Thirteen production systems from 15 states were enrolled in the study (n=188 sow herds). The best machine learning algorithm predicted PRRS occurrences with an accuracy of 78.5%. The 44 biosecurity measures were ranked according to their contribution to the model prediction. The first four most important variables were devotion to breeding genetic replacements, number of swine premises within a three-mile radius, number of breeding females on the premises, and distance to the nearest public road. The probability that a herd had reported an outbreak were higher in farms that had raised breeding animals as genetic replacements. The higher the number of swine facilities within a three-mile radius and the closer the farm was to a public road, the more likely the facility would be expected to break with PRRS. Finally, the PPVs were correlated with the number of reported PRRS outbreaks (correlation of 72%). The majority of farms that had reported no outbreaks in the past five years had low (<50%) probability of having a PRRS outbreak (i.e., low score) while a larger number of PRRS cases were expected for farm who had reported at least one outbreak (i.e., high score), with few exceptions.
The analysis offers a flexible, shortened approach to screen breeding herd’s PRRS biosecurity vulnerability. This study highlights the value of using data to build upon our understanding of biosecurity risk in an operation.
Project #: 20-071 | Principal Investigator: Hiep Vu| Institution: University of Nebraska-Lincoln | Posted: 2/1/2021 | Keywords: ASFV, heat treatment, swine feces, disinfection, virus inactivation
African swine fever virus (ASFV) is the most devastating viral pathogen which can cause up to 100% mortality in domestic pigs. Currently, the virus is affecting many swine producing countries, leading to the approximately 25% reduction of global swine population. Swine transportation plays a major role in spreading infectious pathogens. Thus, developing a procedure for effectively disinfecting animal trailers will help reduce viral spreading. The most effective method to effectively inactivate pathogens in transportation trails is a combination of washing, disinfecting and drying. However, there are not enough washing facilities to wash all trailers between loads of swine. Therefore, we are interested in testing if it is possible to effectively inactivate ASFV in the presence of organic materials (feces, bedding) through the use of thermal-assisted drying and decontamination (TADD) which commonly operates at the temperature between 63°C and 71°C (Dee et al., 2005).
The objective of this project is to determine the optimal baking time and temperature required to completely inactivate ASFV in aluminum surface contaminated swine feces. We tested the inactivation efficiency of contaminated trays under 3 conditions:
• Baking contaminated aluminum trays at 54oC and 63oC for 5, 10, and 15 min
• Power washing the tray surface with water at room temperature followed by baking at 54oC and 63oC for 5, 10, and 15 min
• Power washing the tray surface with water, spray disinfectant (Virkon-S) followed by baking at 54oC and 63oC for 5, 10, and 15 min
Two different methods were used to evaluate the efficiency of the treatments: PCR to detect of viral genomic DNA and virus isolation to detect infectious virus.
In the first condition, swabs collected from contaminated trays at all time-points post incubation at 54oC and 63oC were positive by PCR, indicating that heat treatment could not eliminate viral genomic DNA. On the other hand, swabs collected from contaminated tray at 5 min post incubation at either 54oC or 63oC were negative by virus isolation, indicate that holding ASFV in the presence of feces at 54oC for 5 min is sufficient to inactivate the virus.
In the second and third conditions, only two swabs collected after washing were positive by PCR at high Ct value (e.g. 37.03). These swabs were negative by for virus isolation. Thus, under the conditions of this study, power washing of the trays with water at room temperature was efficiently remove contaminated material from the trays.
Collectively, results obtained from this research provide valuate information for the develop effective sanitation protocols to disinfect animal trailers to reduce the spreading of ASFV.
Project #: 19-236 | Principal Investigator: Nubia Macedo | Institution: Iowa State University | Posted: 1/31/2021 | Keywords: Streptococcus equi subsp. zooepidemicus, swine, experimental model, PCR
In swine, Streptococcus equi subsp. zooepidemicus (SPZ) caused a severe outbreak in China in 1975. In the United States, a genetically similar and hypervirulent SPZ strain caused high mortality outbreaks in sows and feeder pigs in 2019. There is currently no available challenge model in pigs to evaluate the disease process and diagnostics, which would provide meaningful information about the pathogenicity and epidemiology of SPZ in pigs. To address this need, conventional 6-week-old pigs were challenged using hypervirulent SPZ strains and a genetically different, less virulent SPZ strain. Additionally, sequenced SPZ strains were assessed to identify specific virulence markers, and a multiplex RT-PCR assay was designed for SPZ identification and prediction of virulence in swine isolates. Pigs challenged with the hypervirulent SPZ strains developed severe systemic disease, with mortality varying from 50-100%. In contrast, pigs challenged with the less virulent swine strain had minimal clinical signs and no mortality. The colonization and systemic distribution of SPZ were evident in challenged pigs by culture and PCR. This is the first study to experimentally infect and reproduce the disease in weaned pigs with a hypervirulent swine SPZ strain. Furthermore, pathogenicity differences between genetically different swine strains were described. This experimental model will allow for further investigations on the pathogenesis of SPZ in swine and the development of methods of prevention and control of this emerging disease. The newly developed multiplex RT-PCR provides an accurate and timely assay for detecting and monitoring SPZ in swine herds.
Project #: 19-235 | Principal Investigator: Cesar A Corz | Institution: University of Minnesota | Posted: 1/20/2021 | Keywords: monitoring, emerging, PRRS, PEDV, trends
Objective 1: Monitor trends in pathogens incidence and prevalence – PRRSv, PEDv, PDCoV, Senecavirus and central nervous system associated viruses continue to be monitored. The 2019-2020 season fortunately ended with the third lowest PRRSv breeding herd cumulative incidence (24.5%) during the last 11 years of monitoring. During this year we saw a different trend in breeding herd PRRSv prevalence as the proportion of herd staying in category 1 increased and “plateaued” which had not been seen before. PEDv and PDCoV continued to be present at a low incidence level. Mycoplasma hyopneumoniae was included into our monitoring program through a convenient sample of 8 systems. We now have the ability to quickly add new pathogens, which allow us to be prepared in the case of a FAD introduction.
Objective 2: To conduct prospective monitoring of PRRSv sequence evolution and impact – PRRSv sequence acquisition has been stabilized and these are being acquired on a monthly basis. Throughout the year, several participants contacted us to provide outbreak investigation support. A total of 29 sequence analysis were conducted with one of the latest being a virulent 1-4-4 virus. This allowed us to become a neutral third-party curating sequence to identify similarities and putting systems in contact whenever both parties agree.
Objective 3: Develop capacity to capture and analyze movement data – Transport data is acquired actively and has been analyzed. Movement data can be obtained at a granular level allowing for traceability but most importantly, allows the producer to follow the truck in real-time. Transport biosecurity compliance continues to become an achievable goal including every single step between the truck-wash, loading of pigs, unloading and return to truck-wash. Characterization and description of transport data has shown that few transport vehicles come in contact with 1/3 of the farms of the participating production system highlighting an important level of connectivity.
Objective 4: To expand participation of producers to allow for all to be involved – Expansion continues at three levels: sow, boar and growing pig populations. A new production system joined the SHMP during the year. A total of 30 boar studs from 12 participants have been added to our database. The growing pig population continues to grow with 4 companies sharing their growing pig locations. Work is being done towards linking sow-growing pig populations in our database.
Project #: 19-229 | Principal Investigator: Andres Perez | Institution: University of Minnesota | Posted: 11/2020 | Keywords: swine diseases, global surveillance, hazards preparedness, awareness
The expansion of ASF through Asia has raised concerns in the industry, resonating the events of 2013, when a devastating epidemic of porcine epidemic diarrhea (PED) virus caused far-reaching losses. Those events demonstrated the importance of developing systems to provide situational awareness to stakeholders in near-real time, to coordinate actions between government agencies and the industry with the ultimate objective of preventing or at least mitigating the impact of diseases epidemics. The swine industry is vulnerable to the introduction of pathogens, and their variants, from which the US is currently free. We have developed a private- public-academic partnership to support a system for near real time identification of hazards that will contribute to the mission of assessing risks to the industry. Identified hazards were shared monthly with swine practitioners and the government, to help increase the country awareness and preparedness. Ultimately, the system has kept contributing to identify and early detect or create awareness on key stakeholders to support current prevention and mitigation strategies for introduction of foreign pathogens into the US.
Project #: 19-153 | Principal Investigator: Kimberly VanderWaal | Institution: University of Minnesota | Posted: 11/1/2020 | Keywords: prediction, disease, forecast, infection, swine, virus, PEDV, epidemiology
Despite our growing understanding of the epidemiology of viral diseases such as Porcine Epidemic Diarrhea virus (PEDv) and Porcine Reproductive and Respiratory Syndrome virus (PRRSv), a gap remains between the science and the ability of producers to be able to effectively estimate and respond to spatial and temporal variation in risk. Therefore, the aim of this project was to generate farm-level forecasts of PEDv risk that account for recent animal movements, present disease distribution, and environmental factors. Utilizing data captured by the Morrison Swine Health Monitoring Project, we built machine learning algorithms that predict whether a sow farm will break with PED two weeks in advance.
Our forecasting tool was able to detect approximately 20% of the outbreaks in a region (sensitivity), while 70% of the outbreaks our tool said would occur did indeed happen (positive predictive value).The most important factors related to breaks were animal movements both into the sow farm and its neighbors, as well as the PED-status of the origin farm of these movements. Farm density and ambient temperature were also important predictors. We developed this tool using data for a single US swine-dense region and began applying it to a second region of the country, operating with different industry systems and a different environment. This forecasting tool is operationalized for real-time use – since December 2019, partnering systems have received weekly system-specific predictions of PEDv outbreak risk at farm level. These predictions are made two weeks into the future, which allows systems, veterinarians and producers to act in case a high probability of an outbreak is forecasted. This provides an important tool for informed decision-making and coordinated actions of producers and practitioners to control or mitigate the impact of PEDv outbreaks.
Project #: 19-237 | Principal Investigator: Derald Holtkamp | Institutions: Iowa State University | Posted: 10/1/2020
UVC germicidal chambers are used in swine settings to reduce the microbial load on surface items. Chambers, which may be commercial or homemade, are usually constructed so items to be disinfected are passed through from the dirty side (entry/hallway) to the clean side (office/break room).
UVC germicidal chambers are mostly used for small to medium items like lunch boxes, cell phones, small tools, and medications. Food and semen bags can also be passed through the chamber without negative effects. Repeat exposure of plastics to UVC light may lead to a change in the color or smell of the object. Paper and cardboard cannot be disinfected in a UVC germicidal chamber. Larger UVC chambers, or UVC rooms, can be built for larger items.
Project #: 19-211 | Principal Investigator: Gustavo Machado
Institutions: North Carolina State University | Posted: 10/5/2020 | Keywords: swine disease spread, transmission dynamics, disease surveillance, mechanistic modeling
A limited understanding of transmission dynamics is a major obstacle to prevent and control disease spread among swine populations. Forecasting outbreaks before they occur would allow the swine industry to tailor control strategies and improve pig production. Understanding what combination of strategies may be required to reduce between-farm transmission is key to maintain control of outbreaks while minimizing disruptions to pig production. Our objective is to forecast weekly Porcine Epidemic Diarrhea virus (PEDV) outbreaks by generating high-resolution maps to identify current and future PEDV high risk areas, and simulating the impact of control measures. Three epidemiological transmission models were developed and compared: a novel epidemiological modelling framework was developed specifically to model swine populations, PigSpread and two models built on previously developed ecosystems; SimInf (an stochastic disease spread simulations) and PoPS (Pest or Pathogen Spread). Prediction accuracy across models was compared using receiver operating characteristic (ROC) area under the curve (AUC). The models were calibrated on true weekly PEDV outbreaks from three spatial related swine companies. Model outputs had general agreement with observed outbreaks throughout the study period. PigSpread had an AUC of 0.71, SimInf had an AUC of 0.59 and PoPS had an AUC of 0.80. Our analysis estimates that the combined strategies of herd closure, feedback and reinforcement of on-farm biosecurity reduce 14% the incidence of outbreaks in sow and 20% in Gilt Development Units (GDU) when deployed weekly in sow and GDU farms located in risk areas. Accurate forecasts of PEDV spread are feasible to be generated within a decision making timeframe, but the predictability across all models depends on the stage of the epidemic.
Project #: 19-220 | Principal Investigator: Ganwu Li |
Institutions: Iowa State University | Posted: 2/10/2021 | Keywords: diarrhea, phylogenetic analysis, porcine sapovirus, real-time PCR, recombination
A case from the farm experiencing an ongoing problem with piglet diarrhea in the lactation phase for more than 2 years. The pigs exhibited a self-limiting diarrhea starting around 10 days of age, but typically lost 1-2 lbs of expected weaning weight. Histopathological examination of five clinically affected piglets revealed that 5/5 small intestines had moderate villus atrophy, vascular congestion, and lymphocytic infiltration in the small intestine suggestive of an enteric viral infection. Porcine epidemic diarrhea virus (PEDV), porcine deltacoronavirus, transmissible gastroenteritis virus (TGEV), or rotavirus were not detected from small intestines using real-time PCR assays (rRT-PCR). Additionally, there was no significant bacterial growth from the small intestines. Herein, we document the discovery of porcine sapovirus of genogroup III as the cause of the enteritis and diarrhea use of four independent lines of evidence: metagenomics analysis, real-time RT-PCR, histopathology, and in situ hybridization. To our best knowledge, this is the first evidence that SaV likely serves as the sole etiological agent causing enteritis and diarrhea of piglets in the field in the United States. In addition, a highly sensitive and specific real-time RT-PCR for detecting porcine sapovirus of genogroup III was established. Prevalence survey of more than 500 samples from both pigs with clinical diarrhea and clinical healthy pigs suggests that porcine sapovirus III play an important role in causing swine enteritis and diarrhea and rRT-PCR is a reliable method to evaluate the pathogenicity role of porcine SaV.
Project #: 16-250 | Principal Investigator: Orlando Perez | Institutions: National Centre for Foreign Animal Disease, Canadian Food Inspection Agency | Posted: 8/31/2020 | Keywords: pseudorabies virus, variant, highly pathogenic, triplex PCR, detection, differentiation, DIVA
Pseudorabies virus (PRV) causes Aujeszky’s disease or pseudorabies (PR), fatal encephalitis in newborn piglets, respiratory infection in growing and fattening pigs, and reproductive failures in pregnant sows. It establishes a lifelong latent infection in the peripheral nervous system followed by subsequent intermittent shedding of infectious virus. Since 2011, highly virulent PRV strains that are genetically different from the classic PRV strains surfaced in pig herds in China. Availability of a highly sensitive and specific polymerase chain reaction (PCR)-based diagnostic assay for rapid differential detection of PRV variants is critical to prevent huge economic losses to the U.S. and Canadian pork industries if these strains enter North America and cause an outbreak. Here we describe the development and evaluation of a single-tube triplex real-time-PCR assay for differential detection of variant strains of PRV. The assay targets the intergenic region between the US2 and US6 genes in the PRV genome and is highly sensitive and specific and it did not detect other nontarget viruses including related herpesviruses. The clinical specificity and sensitivity of the assay was evaluated using whole blood, serum, tissue and swab samples collected from known negative and experimentally inoculated pigs with either classical (Bristol) or variant (JS-2012 and HeN1) PRV strains. The targeted genomic region of this assay is also deleted in commonly used PRV gE-deleted marker vaccines, and therefore, the triplex assay did not detect viral DNA extracted from two commercial vaccine strains Bartha K-61 and Bucharest. This single-tube triplex assay can be used for routine diagnostics and epidemiological studies for detection and differentiation of classical strains from variant strains of PRV, and as a differentiation of infected and vaccinated animals (DIVA) assay when PRV gE- deletion mutant marker vaccines are used.
Project #: 19-149 |Principal Investigator: Derald Holtkamp, PhD | Institution: Iowa State University | Posted: 8/21/2020 | Keywords: epidemiological outbreak investigations, rapid response corps, Rapid Response to Emerging Disease Program
The need to quickly identify, control, and eliminate a pathogen in an endemic, emerging, or transboundary disease outbreak in the United States is crucial to protect the swine industry from suffering huge economic losses. In August of 2016, SHIC funded Iowa State University to develop the Rapid Response to Emerging Disease Program (RRP). A Rapid Response Corp (RRC) was formed for the program. The RRC is a nationwide network of veterinarians, state animal health officials or representatives, epidemiologists and, when appropriate, federal animal health officials who are trained, prepared and committed to moving within 24 hours of contact to conduct epidemiological investigations when a new transboundary or emerging disease threat occurs. The standardized approach, methodology, forms, and reports developed for the Iowa Pork Producers Association (IPPA) funded PRRS outbreak investigation pilot project, were adapted to develop the RRP. Online training modules, checklists, investigation forms, surveys, and report templates to conduct a rapid response investigation are now available for download on the SHIC website. The objective of this project was to facilitate the continuation of the RRP beyond the current five-year funding stream for SHIC by automating and streamlining the rapid response investigation process through a web application. This proposal included the planning phase to automate and streamline the process. The RRC was maintained at 35 members for the duration of this project. To support the continuation of the RRP, and to provide members of the RRC with additional training, a pre-conference seminar was conducted at the American Association of Swine Veterinarian’s Annual Meeting in March of 2020. The seminar was titled “Conducting effective outbreak investigations: Learning from our mistakes, part 2.” The seminar followed the successful pre-conference workshop offered as training for the RRC members at the same meeting in 2019. The possibility of integrating the RRP into the National Pork Board’s swine business continuity system (AgView®) was explored. Delays in the development of AgView led to the exploration of simpler, more executable approaches. A web-based version of the investigation form used by the RRC members to conduct outbreak investigations has been developed using Qualtrix, which is a commercially available online survey platform. The web-based version of the investigation form will be tested in Vietnam, as part of another SHIC funded study to investigate African swine fever (ASF) virus on farms in Vietnam.
Project #: 19-148 |Principal Investigator: Cassandra Jones | Institution: Kansas State University | Posted: 2/25/2020 | Keywords: porcine, Senecavirus, virus, feed, ingredient, sampling, biosecurity
Environmental monitoring is commonly used in pharmaceutical, human food, and pet food manufacturing facilities manufacturing as an indicator of pathogenic bacteria in the product. A correlation between the presence of Salmonella spp. and Enterobacteriaceae within feed mills has been demonstrated, but little information is available on how the presence of Enterobacteriaceae (EBAC) correlates with viral pathogen presence, especially on farms or in feed mills. The purpose of this study was to identify Enterobacteriaceae presence in the feed manufacturing facilities of a multi-farm system experiencing a viral outbreak as a method of identifying biosecurity gaps.
Project #: 19-219 | Principal Investigators: Derald Holtkamp, DVM, MS | Institution: Iowa State University College of Veterinary Medicine | Posted: 12/2019
The swine industry has focused much of its efforts on improving biosecurity in breeding herds; while little attention had been paid in wean-to-finish growing sites. One risk event that has the potential to introduce virus into grow-finish pigs is load-out during marketing. In order for the remaining pigs in the group to become infected during load-out, viral contamination must be transferred from the contaminated livestock trailer, driver or other carrying agents to the pigs in the barn. Unfortunately, little research has been done to assess how frequently this occurs or to assess alternative biosecurity measures to reduce the frequency.
Project #: 19-236 | Principal Investigators: Nubia Resende-De-Macedo | Institution: Iowa State University | Posted: 3/24/2020 | Keywords: bacterial pathogens, emerging diseases, veterinary epidemiology
High mortality events due to Streptococcus equi subspecies zooepidemicus (Streptococcus zooepidemicus) in swine have not previously been reported in the United States. In September and October 2019, outbreaks with swine mortality up to 50% due to S. zooepidemicus septicaemia were reported in Ohio and Tennessee. Genomic epidemiological analysis revealed that the eight outbreak isolates were clustered together with ATCC 35246, a Chinese strain caused outbreaks with high mortality, also closely related to three isolates from human cases from Virginia, but significantly different from an outbreak-unrelated swine isolate from Arizona and most isolates from other animal species. Comparative genomic analysis on two outbreak isolates and another outbreak-unrelated isolate identified several genomic islands and virulence genes specifically in the outbreak isolates only, which are likely associated with the high mortality observed in the swine population. These findings have implications for understanding, tracking and possibly preventing diseases caused by S. zooepidemicus in swine.PCV3 may cause death in fetuses and myocarditis and systemic vasculitis in pigs.
Project #: 18-201 | Principal Investigator: Albert Rovira | Institution: University of Minnesota | Posted: 3/30/2023 | Keywords: PCV3, abortion, myocarditis, vasculitis, diagnostic laboratory, lesions, clinical signs
Porcine circovirus type 3 was discovered in 2016 in the US associated with cases of systemic disease and reproductive disorders. Multiple studies performed during the last two years have shown that this virus is widespread and has been around for decades. It can be found in multiple tissues and samples, in pigs with multiple clinical conditions and in healthy pigs. However, we are lacking precise information on the relevance of this virus and its potential to cause disease in pigs. The objective of this proposal was to mine the diagnostic data obtained by the MN VDL during the last 2 years to identify associations between the presence of PCV3 and its viral load and specific lesions and clinical conditions. PCV3 results, clinical signs and information on the lesions for each pig were retrieved from the MN VDL laboratory information management system, submission forms and diagnostic reports. PCV3 frequency in pigs with different clinical signs ranged from 12% to 27%. No significant associations were observed between clinical signs and the presence of PCV3. In PCV3-positive pigs, no clinical signs were significantly associated with having a higher load of PCV3. PCV3 frequency in pigs with different lesions ranged from 0% to 62%. The only lesion that had a significant association between its presence and PCV3 infection was heart vasculitis/perivascultis. In PCV3-positive pigs, higher viral loads were significantly associated with pigs with myocarditis, heart vasculitis/ perivasculitis, kidney vasculitis/perivasculitis and dermatitis. This study did not identify any significant associations with clinical signs. However, the presence of PCV3 in 20% of fetuses is remarkable. In addition, the samples with the highest PCV3 concentration in this study were from fetal tissues. Lesions of myocarditis and systemic vasculitis were associated with the presence or the amount of PCV3 in tissues. The lack of significant results for other lesions does not exclude the possibility of a real association and may be due to confounding factors or limited data. In summary, this study provides an objective view of the relationship between PCV3 and disease, based on a large dataset of diagnostic cases. PCV3 is a very common virus that circulates in healthy populations and can be detected in around 20% of the pigs submitted to the diagnostic laboratory. Therefore, it is important to differentiate when PCV3 plays a significant role and when it does not. The results from this study support previous studies that suggested that PCV3 may cause death in fetuses and myocarditis and systemic vasculitis in pigs.
Project #: 20-072 | Principal Investigator: Scott Dee | Institution: Pipestone Applied Research | Posted: 5/11/2020 | Keywords: animal feed, feed mitigation, ice-block challenge, swine viral diseases
In 2014, contaminated feed was identified as a vehicle for the transport and transmission of PEDV. Over time, similar conclusions were drawn regarding the role of feed and the risk of African swine fever virus, Senecavirus A (SVA), Pseudorabies virus and Classical swine fever virus. However, as these results were based on laboratory studies, our goal was to validate these observations using a real-world “demonstration project” approach, simulating the actual transport of feed contaminated with SVA, PEDV and PRRSV across the United States in a commercial transport vehicle. Samples of conventional soybean meal, organic soybean meal, lysine, choline and vitamin A were spiked with all 3 viruses and placed in a trailer of a commercial transport vehicle. The samples were stored in vented containers to allow exposure to environmental factors including temperature and relative humidity during the trip. Overall, the journey involved 21 days, 107 hours of transport, crossed 14 states and covered approximately 6000 miles. At the end of the study period, samples were tested for the presence of viral genome by PCR and viable virus by swine bioassay. Results indicated the presence of viable PRRSV, SVA and PEDV in both soy products, while viable SVA was recovered from all 5 ingredients. In contrast, survival was limited in the vitamins and amino acid ingredients. These results validate published laboratory data indicating the protective nature of soy-based feed ingredients and demonstrate that certain viruses, such as SVA are very stable in feed. In closing, this novel approach did demonstrate that three significant viral pathogens of pigs could survive in select feed ingredients during an actual shipping event, involving diverse environmental conditions and realistic transit periods. It is hoped that the information derived from this study will help to unify opinions across the swine industry, the veterinary profession, and governmental agencies regarding the risk of feed.
Project #: 16-262 | Principal Investigator: Rodger Main | Institutions: Iowa State University Veterinary Diagnostic Laboratory, Clemson University, Kansas State University Veterinary Diagnostic Laboratory, South Dakota State Animal Disease Research and Diagnostic Laboratory, University of Minnesota Veterinary Diagnostic Laboratory, USDA NAHLN | Posted: 8/13/2018 | Keywords: data, diagnostic, HL7, LOINC, NAHLN, standard, swine, VDL
Seamless integration of diagnostic data from any number of veterinary diagnostic laboratories (VDLs) into third-party database applications for further analytical and reporting purposes has long been recognized as a critical element necessary for proficient detection, monitoring, response, and/or management of significant diseases across a region, state, or nation. Establishing and adopting the use of universally recognized diagnostic data standards and a common electronic messaging schema are foundational elements necessary for the development of sustainable and scalable systems of connectivity and web-based analytical tools necessary to support the current needs and future demands of the US Pork Industry.
This highly collaborative initiative served to further develop the core infrastructure and diagnostic data standards necessary for US Pork Industry stakeholders to effectively harness the capabilities that any number of existing or yet to be developed (web-based) animal health information management technologies aim to provide. Primary deliverables included the development of a more comprehensive electronic message schema; a greatly expanded formulary of now more than 700 standardized test result codes covering the full-spectrum of diagnostic tests commonly conducted on swine case submissions to veterinary diagnostic labs (VDLs); an intuitive, web-based search engine that enables users (e.g., VDL information technology personnel) to readily identify the appropriate diagnostic test result code(s) (i.e., Logical Observation Identifier Names and Codes, or LOINC®) to use for each of the specific assays conducted and reported from their laboratory; and a web-based electronic message validator that allows VDLs to test, trouble-shoot, and validate their electronic messaging capabilities.
Each of the VDLs collaborating on this infrastructure development project demonstrated their ability to electronically synthesize and successfully deliver results from diagnostic case submissions utilizing the updated electronic messaging schema and expanded formulary of diagnostic test result codes. To ensure national level scalability, the updated electronic messaging schema derived from this project is consistent and fits within the overarching architecture of the Health Level Seven® (HL7) messaging schema adopted by the United States Department of Agriculture National Animal Health Laboratory Network (USDA NAHLN). The expanded formulary of standardized codes for diagnostic test results (LOINCs); the updated and more comprehensive HL7 electronic messaging schema; the standardized diagnostic test result code (LOINC®) search engine; and the HL7 electronic message validator application developed as a result of this project will be made available for use by VDLs across the USDA NAHLN.
The infrastructure developed in accordance with the primary deliverables of this initiative will provide a lasting foundation for enhancing the adoption and use of universally recognized diagnostic data standards and HL7 electronic messaging capabilities in swine interest VDLs in the USDA NAHLN. Furthering the establishment and use of such standards are critical building blocks for enhancing the connectivity and interoperability of diagnostic information across VDLs and for broadly transcending swine diagnostic information management into the digital era. Albeit these developments centered at the four VDLs collaborating on this project and were specifically focused on diagnostic tests conducted on US swine, this precedent-setting work could readily be expanded and emulated for use across any number of animal species and VDLs.